Irwin Rose
Biographical

We left my birthplace, Brooklyn, New York in 1939 when I was 13. I enjoyed the ethnic variety and the interesting students in my public school, P.S. 134. The kids in my neighborhood were only competitive in games although unfriendly gangs tended to define the limits of our neighborhood. The major extracurricular activities that I can remember were a Victory Garden on school grounds, our contribution to the war effort, and a favorite sport, handball, played between the walls of our apartment house.
Mother, Ella Greenwald, was an American born into a family that included one sister and four brothers, all born in Hungary. Father, Harry Royze, had two brothers and a sister from the Odessa region of Russia. The Greenwalds and the Roses were secular Jews and the children more so although my younger brother and I spent some time in Hebrew school to please Grandfather Rose.
Due to my brother having rheumatic fever the family was advised to go to a high and dry climate, Spokane, Washington, where my mother’s sister had a comfortable home that could accommodate us. This left my father behind tending his flooring business, an arrangement that I never understood and felt conflicted about. Father’s visits were few and far between. The war was going on. Mother did secretarial work in the Navy Supply Depot in Spokane while we kids were making our way through the Spokane school system.
I worked during the summers at a local hospital, chiefly helping out in the psychiatric ward. In time I came to see myself following some career that involved solving medical problems. No one in my family had followed a career in research. Uncle Arthur G. was an excellent violinist and artist, and taught cabinet making at a trade school in Brooklyn. Uncle Dave R. would have become a lawyer had the economic depression not led him into the U.S. Internal Revenue Service. There was no one in my circle from whom I could expect to get advice.
Initially, I thought problems on how the brain works to be the most interesting. But it was necessary to be practical, and concentrate on less obscure matters when I entered Washington State College. Besides, there were no courses given in neurobiology. However, I was strongly influenced by Prof. Herbert Eastlick, who urged his zoology students to set high standards for themselves, and then proceeded to the University of Chicago after a brief period in the Navy. My PhD thesis problem was to determine if the DNA content of rat tissues increased if there was B12 in the diet. This problem was suggested by my adviser based on the observation that thymine could replace vitamin B12 in a lactobacillus. I analyzed the DNA of tissues of rats fed with diets that varied in B12. This project was doomed to failure when the genetic nature of DNA was revealed, and I found that the DNA content per cell of liver was independent of diet1.
PhD Work. I had to think of a new thesis project. Anxious to make up for lost time, I picked a problem out of my freshman biochemistry lecture notes. The Putnam/Evans group was interested in determining the origin of the nucleic acid components of bacteriophage synthesized in E. coli and Frank Putnam’s lectures described experiments of Hammarsten, Reichard, and Saluste2 as background information. 15N-cytosine, the free base, had been found not to be incorporated into DNA although 15N-cytidine was incorporated into rat liver DNA. It was obvious for me to ask if there might be direct utilization of the whole of cytidine, ribose and all, in the biosynthesis of deoxycytidine. That would be a shock. I learned from Peter Reichard, during a 2004 meeting in Stockholm, that the export to Sweden of 14C-compounds was forbidden by the U.S. Atomic Energy Commission at that time, otherwise they certainly would have done the obvious follow-up experiment, using uniformly U-14C labeled cytidine themselves.
I made RNA from Euglena gracilis grown on 14CO2. I had to work out the determination of the independent specific activities of the sugars and bases which I did by treating the nucleosides with nucleoside phosphorylase and hypoxanthine to exchange for the base to be analyzed. Then by paper chromatography, using a medium containing borate to retard the migration of ribosides, I could also isolate deoxyinosine and cytosine. Although U-14C cytidine did not label the deoxyribose of E. coli DNA, I found the deoxycytidine of DNA of rat organs to be almost uniformly labeled. The 14C content was far in excess of the negligible radioactivity in the purine deoxynucleotides3. Therefore by both criteria it appeared certain that the 14C reached the deoxyribose directly from the cytidine. Reichard repeated and extended this experiment with U-14C uridine in 1957 with much the same result for the deoxycytidine and thymidine4.
It would have been reasonable for me to try to work out the enzymology of ribonucleotide reduction after graduating. Peter Reichard at Yale on a post-doc from Sweden, asked me about my intentions. But I was not anxious to take on a heroic problem at this early point in my career. I was interested in learning more about the principals of enzymology.

Stereochemistry at Chicago. Ogston’s 1948 paper proposing, in effect, that the ability of an enzyme-substrate complex to distinguish between identical groups on a tetrahedral carbon was a consequence of the asymmetry of the complex5, was a matter of hot debate in chemistry/biochemistry circles at Chicago in 1950 where the enzyme was still a black box and the emphasis was on the chemistry of changes in the substrate. In particular the Ogston idea could justify the conclusion in the experiments of Myron Bender, done in Chemistry at University of Chicago, that the absence of back labeling of an ester in 18O-water during enzymatic hydrolysis could not rule out a tetrahedral intermediate. Bender had already shown that back labeling occurred during ester hydolysis in alkali6. In the case of the enzymatic reaction based on Ogston one would expect to lose all the 18O on stereospecific return of such an intermediate to the ester. These thoughts morphed into the positional isotope exchange idea in 1976.

Thus I became challenged to establish the absolute stereochemistry of enzymatic reactions and determine its mechanistic significance, if any. This did not seem such a formidable task, although it was not until 1963 that Kenneth Hanson and I solved the historically important problem of the prochirality of citric acid7, which was necessary for me to gain a proper perspective on the aconitate hydratase reaction.
Yale. In 1955, after post-doctorals at Western Reserve University with C. E. Carter and at New York University with Severo Ochoa, I was fortunate to be invited by Joseph Fruton to become an Instructor in Biochemistry at Yale University Medical School. The first year at Yale was notable for the following three developments. Not willing to spend the time it would take to get the Department’s mass spectrometer working, I turned to the scintillation counter that was available in the medical school lab of Seymour Lipsky, an M.D. with a passion for exploring and exploiting new methods. One of the pioneer instruments to become available came from a small start-up company in New Haven that Lipsky had been encouraging, the Technical Instrument Company. Lipsky also had a sample of tritiated water which together with his counter got me started on experiments I wanted to do.
A second important event of my first year at Yale was to learn from Mel Simpson of his paper showing an apparent ATP requirement for protein breakdown in a liver slice system. This observation required further study which I attempted on the side for the next twenty years.
But the crowning event of 1954-1955 was my proposal of marriage to Zelda Budenstein, a graduate student in the Department. Fortunately, I caught up with her before she graduated. Her mother, widowed since Zelda was age five, came to live with us. She was much loved and a great help with the four children that were in our onrushing future. She enabled Zelda to have a research career, often paralleling mine, which she continued until 1987 when she retired to devote full time to her peace and social interests.
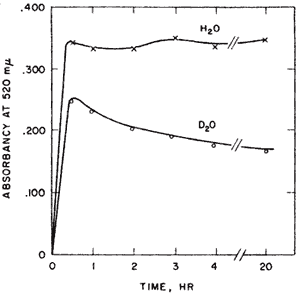
Aldose-ketose isomerases. Probably the most interesting experiment of my nine years at Yale, l954-1963, was interesting from the way it developed and the confidence it gave me that I might be able to do research after all. I had been looking for evidence of proton transfer in enzymes that catalyzed aldose-ketose interconversions. We had been mistakenly unsuccessful in not finding the small amount of transfer that was later detected in the triose P isomerase reaction. Y.J. Topper had reported that glucose 6-P isomerase in D2O formed glucose-6-P (G6P) containing about one deuterium using crystallization of the barium salt as a G6P trap8 suggesting that there might be no proton transfer between reactants of this enzyme. The importance of showing some transfer would be that it would provide a clue to the catalytic process. Complete transfer would suggest a hydride transfer. Complete exchange would suggest a carbanion intermediate but would not implicate the enzyme as the base. However, the occurrence of both transfer and exchange would result if the abstracted proton were to exchange to some extent before a second proton transfer. Such a result would imply a single base mechanism. No such result had yet been reported. In unrelated experiments I observed a puzzling phenomenon with G6P isomerase. When G6P was used with isomerase in D2O the colorimetric analysis for fructose-6-P passed through a maximum before reaching a final value. This overshoot of equilibrium that occurred only in D2O, Figure 1, was very puzzling.
Professor Julian Sturtevant of the Chemistry Department sagely asked me if I was sure of my assumptions. I soon figured out that Topper’s experiment might have been misleading. Perhaps his barium trap of G6P was not good enough. If there were both transfer and exchange in D2O then as the product returns to the enzyme there would be another opportunity for exchange until the product becomes fully exchanged. The fructose-P would go from a partly H- form initially, to an all D-form in the exchanged position at C-1. Now the only thing necessary for my strange result to make sense was to find an isotope effect in the color reaction for ketoses in acid. This was shown by finding the equal amounts of fructose-6-P at 20 hrs when assayed by a different method9.
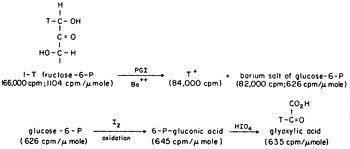
We could show transfer using 1-T fructose-6-P with the isomerase using Ba to form the crystalline salt of G6P as it formed in the presence in a small amount of seed crystals, Figure 2. The 6Pgluconate made from the G6P was only labeled at C-2, the site to which transfer had occurred. We also showed that transfer was between carbons of the same molecule and that the extent of transfer was greatest at low temperature, indicating that the greatest effect of heat was on the dissociation of the intermediate E-T. The proposed intermediate, EH.enediol-P was found to partition about equally in the forward and back direction as shown by the equal incorporation of T-water into product and substrate at early times. A low specific activity of the product was consistent with the slow exchange of the intermediate which would also be expected to show some discrimination against T of the solvent. Since only one T entered the C-1 position of the fructose-6P at equilibrium in tritiated water the abstraction and exchange with solvent were stereospecific. The absolute stereochemistry of 1-2H-fructose-6P was rigorously established by neutron diffraction crystallography as suggested by Lindo Patterson of Fox Chase using the 6Li salt of monodeuteroglycolate10. This was the first example of the use of neutron diffraction to establish the absolute configuration of a molecule made asymmetric by isotopic substitution.
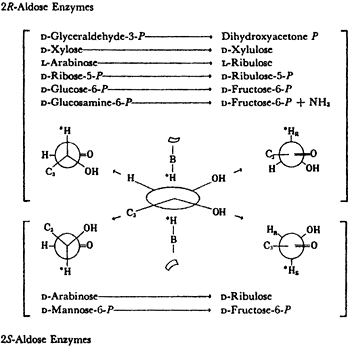
All isomerases use cis-enediols intermediates. As shown in Table 1, isomerases that are specific for 2R-aldoses were found to activate the 1-R proton of the ketose produced. Isomerases specific for 2S-aldoses produce ketoses that exchange their 1S proton11. For these proton transfers to have been intramolecular, they would both have come from the same face of the enediol intermediates. This implies a cis-enediol intermediate. cis-enediol intermediates have the advantage of requiring only a single electrophile to polarize the carbonyl of either substrate. The requirement for one base and one electrophile may be a major factor in preserving the cis-enediol mechanism in evolution.
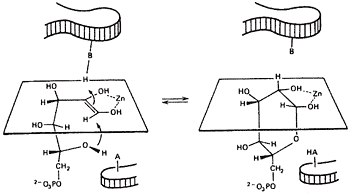
A relation between the cis-enediol mechanism and the anomeric specificity of isomerases. The open chain forms of sugars are minor species in aqueous equilibria. Therefore a ring opening step will be the first step catalyzed by isomerases for any pentose or hexose substrate. Since ring closing is the reverse of ring opening, we wondered if enzymes with a cis-enediol intermediate would be defined in the ring closing step. Ring closing should occur from the face opposite that used by the proton donor. For example, if the proton approaches C-3 from above the plane, the C2-C3 bond will become oriented below the plane and the C5OH must approach the C-1 carbonyl that is being generated to form the cyclic product from below, see figure 311-13.
The anomeric specificities of 5 isomerases were determined in the course of our studies on the isomerases that continued until 1973. The results were as predicted, see Table 2. Thus a-xylose were the substrates for the 2R-aldose enzymes in their production of fructose 6P+NH3 and xylulose, respectively, and b-L-arabinose and B-D-mannose-6-P were the specific substrate for the 2S-aldose isomerase.

The specificity of glucose-6-P isomerase is unusual. Consistent with the above considerations a-G6P and a-F6P are the favored substrates. However, isomerization of b-G6P occurs at a significant rate. It has been proposed that the open chain aldose while on the enzyme undergoes torsional inversion at the C2-C3 bond before ring closing. This kind of motion, if it were to occur in the ene-diol of G6P could add the C-2 proton to the flip face and produce mannose-6-P. Seeholzer has reported the activation of the C-2 proton of mannose-6-P by G6P isomerase14.
A nomenclature for ring face designation15. In looking for a way to designate the ring face of a simple ring compound such as G6P or of the two rings of a nucleoside, or of fused ring compounds such as sterols, we realized that a universal method could be devised using the established rules for numbering the atoms of rings as contained in standard chemical handbooks15. A face is considered a if the numbered ring atoms are seen to increase in a clockwise direction, otherwise this face is designated the b-face. The method is not subject to changes in ring substituents as is the D/L naming of sugars. One can readily communicate structural information without resorting to pictures by this method. For example, noting that in Watson-Crick base pairing the paired bases have the same face-orientation in forming the antiparallel double helix tells us much of what we need to know to build the correct structure.
Fox Chase. In l963, we moved to The Institute for Cancer Research of the Fox Chase Cancer Center in Philadelphia. The Institute was a unique place, in my experience. Since its founding it was governed by the notion that to understand cancer one needed a broader understanding of biological science. In its first expansion from center city in 1949, its faculty included crystallographers, embryologists, chemists, biochemists and medical biologists without departmental restrictions. I was attracted by the chance to learn from a wide range of researchers attending each other’s seminars as well as the freedom from teaching. Research support came from our own competitive grants but the Institute had a generous history of tiding you over. We were not required to go after our own salaries which came from an NIH core grant, the first of its kind, which also supported institutional facilities. Decisions were made by the Director, Timothy R. Talbot, M.D. with the advice of a group of the staff and eminent outsiders. Zelda had a lab and grant of her own. She quit science to be more active in the Nuclear Freeze and other peace efforts in 1987. In 1977 when Avram Hershko requested to share our lab space and facilities, he and his group were welcomed for the 22 years he was with us, either on sabbatical or during summers.
Whole cell systems. In the next few years I started a series of investigations using isotopes to examine problems of metabolism. It had been observed by Harland Wood’s group at Western Reserve that when 14C-lactate or 14C-glycerol are fed to a fasted animal the labeling of the glucose units of liver glycogen was such as to suggest that triose-P isomerase had failed to equilibrate its two triose phosphate substrates during synthesis, or that the condensation reaction of FDP aldolase did not give equal labeling of the two halves of FDP. We had earlier investigated this latter, using isotope exchange at equilibrium (possibly for the first time) showing that the C456 of FDP exchanged more rapidly than C12316.
Asymmetric labeling due to incomplete equilibration at the triose-P isomerase step seemed unlikely given the enzyme’s high efficiency. This question was settled by showing, with Wood’s group, that the asymmetry obtained with 14C-labeled glycerol in which a deuterium was present in the position abstracted in the isomerase reaction was greater in the direction predicted by incomplete equilibration of that enzyme17, an expression of the primary isotope effect of the isomerase reaction that we had shown previously.
Yale graduate student Bob Kemp determined the fates of reduced pyridine nucleotides, NAPDH and NADH in Leuconostoc mesenteroides fermentation and growth using 1-T glucose, 1-T 2 deoxy-glucose and 3-T glucose. Preferential use of 1-T for ethanol production and 3-T for lactate production indicated partial separation of dehydrogenase products18. NADH derived from glucose 6P dehydrogenase which also oxidizes 2 deoxyglucose-6-P was the primary source of reducing equivalents for lipid brosynthesis.
Control of glycolysis. In the following years we reported studies on the regulation of glucose degradation in human red blood cells. Glucose transport is rapid so that the first irreversible step, hexokinase, must determine the rate of net flux. 14C-glucose utilization was shown to be inversely related to the G6P level over a 40 fold range using methylene blue and inosine to vary the steady state G6P, even as many other metabolites varied greatly without showing effects on this linearity19. Thus any condition that affects the rate of glucose utilization of red cells must do so by either having an effect on G6P or on the activity of the hexokinase itself. A remarkable effect of Pi in stimulating red cell glycolysis was traced to its interference with the binding of G6P to the enzyme without itself having an effect on the hexokinase rate20.
Using measurements of isotope exchange rates we were led to conclude that the accumulation of triose-P intermediates in human red cells that were incubated with high levels of orthophoshate was due to equilibrium and not a rate limiting step. The low levels of NAD and pyruvate caused by net synthesis of 2, 3-bisPglycerate shifted the equilibrium of the glycolytic intermediates toward high triose-Ps21.
Glucose-1, 6-P2 of the brain. Lowry et al. reported the rapid depletion of the normally significant amount of G16P2 during mouse brain ischemia. We have been particularly interested in the role this compound might play in brain function. We purified an enzyme that used G1P with glycerate-1,3-P2, as the phosphate donor, not ATP or FDP22, 23. Mg was a required confactor but Zn was equally active and much more tightly held. About 65% of the activity of a fresh brain extract resistant to EDTA is probably the enzyme in the Zn-form. The synthetase is strongly inhibited by concentrations of citrate and FDP that are found in the brain suggesting a regulatory role of G16P2, and that its synthetic rate may be different in different regions of the brain just as its distribution was shown to be localized24.
Zelda discovered a brain-specific G16P2 phosphatase with an absolute requirement for inosine-5-P25. Since IMP is the major product of ATP breakdown in the ischemic brain this strongly points to activation of the phosphatase as the cause of the rapid fall in G-1,6-P2 in ischemia.
Mitochondrial hexokinase. A problem that warrants further study derives from our observation in 1967 that instead of being in the cytosol most of the hexokinase of tumor and animal cells is associated with the outer membrane of the mitochondrial fraction26. It has recently been reported that the release of mitochondrial hexokinase and some associated proteins may play a role in the onset of the apoptosis cascade27.
The mechanism of synthetases. When I felt that we understood control of the red cell glycolytic system my attention returned to mechanism questions: ATP driven synthetases, reactions such as glutamine synthetase (gluamate + NH3 + ATP –> glutamine + ADP + Pi) may be written to go in three steps: ATP + E -> ADP + E~P, E~P + glutamate –> E. glutamyl~P, E. glutamyl~P + NH3 –> -glutamine + Pi28. However, no ADP/ATP isotope exchange could be found unless all of the reaction components were present. On the other hand, Meister had performed a pulse/chase experiment that seemed to suggest the formation of an activated intermediate. The enzyme, pulsed with ATP and 14C-glutamate, was chased with unlabeled glutamate plus hydroxylamine. 14C-glutamyl-hydroxamate was found in the chase. However, one could argue that a non-covalent complex of E.ATP.glutamate may not have lost the labeled glutamate more rapidly than the chemical reaction would have occurred when the NH2OH entered the complex. A similar assumption was made by Jacob bar Tana, who I met at the Hadassah Medical School in Jerusalem on our sabbatical in 1972. He was having trouble with irreproducible results with phosphofructokinase testing for an E~P mechanism by preincubating the enzyme with ATP32 and chasing it with a mixture of F-6-P and unlabeled ATP. I suggested that he might be capturing E.ATP rather than E~P.ADP. We agreed to examine the assumption that dissociation of a binary complex must be faster than chemistry on bar Tana’s next visit to the U.S. We used yeast hexokinase with 14C-glucose (the pulse) followed by ATP plus a large excess of unlabeled glucose (the chase). Labeled G6P was indeed found in Michaeli’s proportion to the ATP in the chase up to the full measure of enzyme occupied by glucose in the pulse29. Dissociation of glucose was indeed slower than its phosphorylation contrary to the generalization that chemistry is slow. Thus was demonstrated the first pulse/chase experiment from which could be determined the functionality of binary complexes, their rate of dissociation and the extent of dissociation of glucose from the ternary complex.
Pulse/chase variants. Subsequently, our lab used the pulse/chase method to study hexokinase in the steady state by comparing the 14C-G6P formed in an acid quench with that formed in a high substrate chase with time of quenching, a procedure that gave the position of the central equilibrium, ~1, and the rate constants for all interconversions30. We did pulse/chase experiments in which the labeled glucose came from hexokinase in the crystalline state either alone or together with ADP31. Although the crystals dissolve slowly the results suggest that their form at that time reflects the form in the crystal. Crystals with glucose alone required the same amount of ATP for 50% trapping as did E.glucose in solution. On the other hand E.glucose.ADP grown crystals from which the ADP had been removed by washing required much less ATP. We also used standard pulse/chase to find that 4 atoms of tritium, with the specific activity of water in the pulse have positions on the enzyme aconitate hydratase, probably an arginine-threonine pair in the active site that could be trapped by the cis-aconitate in the conversion to citrate32.
Positional isotope exchange (PIX). Although traditional ATP/ADP exchange as a test of E~PO3 or E-glutamyl~PO3 intermediates in the glutamine synthetase reaction would fail if the ADP did not leave the enzyme a different kind of isotope exchange might be observed, one in which the beta-gamma bridge oxygen and the beta-nonbridge oxygens of reisolated ATP had mixed with each other. This would only require the phosphoryl oxygens of the bound ADP intermediate be capable of undergoing positional isotope exchange, depending on a symmetrical torsional motion before returning to ATP. This new exchange process was observed in a cleverly designed experiment done by post doc, Fred Midelfort33. The PIX rate in the presence of glutamate was consistent with the reverse rate of the enzyme. The exchange rate was decreased by NH4+ consistent with a decrease in the concentration of E.glut~P.ADP complex.
Recycling. More recently, we have become interested in how an enzyme recovers after product formation. Changes in the active site of an enzyme that occur when a substrate is converted to product must be reversed before the reaction can occur again. With rare exception, this recycling of the active site occurs before the product has left the enzyme and is therefore counted as part of the product-off step. Are recycling steps ordered or random? Does the sequence replicate the order of steps during the reaction itself? Fumarase has several properties that mark it as a rare example in which, when salt is present to accelerate the release of a product, a further sequence of steps remains that is rate limiting for the whole reaction cycle. Distinctive enzyme isoforms become identifiable as shown by the occurrence of noncompetitive inhibition by product analogues34, 35. Added labeled reaction product rebounds to substrate when unlabeled substrate is added showing that the product form of the enzyme is slow to recycle, the Britton counterflow effect36, 37. D2O and glycerol show uncompetitive inhibition patterns indicating that their effects occur after an irreversible step, product release. Noncompetitive inhibition is rarely seen in enzymes that catalyze reactions requiring only one substrate. When it is seen, it is an indication of slow recycling.
The chemical interconversion of fumarase reaction, a carbanion mechanism, is the most rapid part of the reaction cycle. Thus at equilibrium, 18O-water exchanges into malate more rapidly than does fumarate. Perhaps the details of the reaction chemistry can be determined from the details of the recycling. That this seems to be the case is indicated by the pattern of inhibition. The carbanion intermediate analog, 2-hydroxy, 3-nitropropionate is competitive with respect to both malate and fumarate and therefore represents the intermediate species in the recycling whereas all other inhibitors are competitive with only one or the other of the substrates and therefore represent product species.
The ability of salt to increase the rate of product release is lost when the basic amino acid residues that reside between the active site and the solvent are mutated to neutral forms38. The mutated enzyme is now rate limited by the product-off step, not recycling. We speculated that the negatively charged products are released through a positively charged channel that is made more rapid by salt.
Enol-pyruvate. Heavy atom NMR spectroscopy of the hydrogens of P-enolpyruvate led to Mildred Cohn’s solution of the reaction stereochemistry of the reaction of enolase using (3R)-phosphoglyceric acid-3-d39. This led us to the solution of many reaction stereochemistries with enzymes using specifically labeled 3D, T-PEP40.
Our longstanding belief that enolpyruvate is the intermediate in the pyruvate kinase reaction was confirmed by direct chemical analysis of pyruvate kinase in the steady state41. We could also show that enolpyruvate synthesized by treating PEP with a phosphatase was used by pyruvate kinase at an appropriate rate although its affinity was less than expected for an intermediate42.
Other problems. In a surprise result, the Schiffs bases of dihydroxyacetone-P and of FDP were found to precipitate with the protein of muscle aldolase in cold acid43. This property due to the stability of the Schiffs base intermediates in acid and the acid lability of the eneamin-Ps were used to determine their concentrations as intermediates in the aldolase reaction44. Ring formation of the adducts was indicated. Thus 5-deoxyFDP formed a much less stable acid precipitate than did FDP.
The interconversion of citrate and isocitrate by aconitate hydratase was shown to occur with complete retention of the proton transferred between C2and C3 but with no retention of the OH- between C3 and C2. The bound cis-aconitate intermediate must flip over, probably around a CH2COO- pivoting on the ferrous iron of the active site so that the enzyme bound proton can approach from the opposite faces of the intermediate in generating either of the products45.
Aconitate isomerase catalyzes the interconversion of cis- and trans-aconitate by a 1, 3 allylic rearrangement that uses the (pro-S) hydrogens of the two substrates. The stereochemistry and the transfer of tritium are consistent with a single base carbanion mechanism46.
The physical separation of H- from D-labeled molecules made possible by their different rates of reaction was used to establish that the transfer of hydrogen in G6P isomerase was intramolecular9 and that the site of attachment of CO2 to ribulose-1, 5-P2 to produce two molecules of phosphoglyceric acid had to be to C-2 rather than C-4 in the RUDP carboxylase reaction47.

In this chapter I have mentioned most of the problems that have engaged me and my coworkers until the time of my retirement in 1997 with the exception of ubiquitin related studies that are summarized in the next chapter, “Ubiquitin at Fox Chase”. Following my retirement, Zelda and I moved to Laguna Woods in Southern California where I was able to continue lab work using space and facilities shared by Ralph Bradshaw at nearby U.C. Irvine.
During this period Dr. James Nowick of Chemistry and I examined the mechanism of methylglyoxal synthetase (dihydroxyacetone-P -> methylglyoxal) trying to understand our observation48 that the methyl group of the product was formed nonstereospecifically. We could show spectroscopically that the enol-aldehyde of methylglyoxal was really the enzyme’s product49. Ketonization occurs off the enzyme and therefore is nonstereospecific. Ketonization to methylglyoxal is very slow but is assisted by thiols which interrupt the double bond conjugation by formation of a thiolhemiacetal. It was perhaps for this reason that the next enzyme, of the sequence, glyoxalase I, has the adduct of methylglyoxal with glutathione as its substrate and D-lactyl – glutathione as its product.
We concluded our paper with the observation that organisms that had no glutathione would have to evolve another mechanism to dispose of methyglyoxal which is toxic to cells due to its action in cross linking macromolecules. Our most recent study shows that the mycobacteria that have mycothiol, (N-acetyl cysteine-glucosamine-inositol), as its major reducing compound have indeed evolved an enzyme that carries out the same kind of sequence with a “glyoxalase” that produces lactyl-mycothiol (unpublished). This is therefore a second example in which an enzyme uses the non-enzymatic product of a previous step as its substrate.
Looking back on my 50 year eclectic journey in research, I am grateful that it has gone as well as it has, although still not clever enough to open the black box of enzyme structure. The approach I have taken was successful, in the least, in attracting outstanding postdocs, some of whom were on hand in 1975-1980 when with Avram Hershko, we pursued the ubiquitin/protein breakdown work.
I am especially grateful to those who have helped me find solutions to the many problems that troubled us in the early days of biochemistry. Among them were Jacob bar Tana, Steve Benkovic, J. F. Biellmann, Mildred Cohn, Aaron Ciechanover, Don Creighton, Raj Gupta, Arthur Haas, Kenneth Hanson, Avram Hershko, Anthony Jaworowsky, G. Kaklij, Robert Kemp, Judith P. Klinman, David Kosow, Donald Kuo, Gustav Lienhard, Oliver Lowry, Alton Meister, C. Fred Midelfort, Gerd Mullhofer, Koko Murakami, James Nowick, Edward O’Connell, Frank Oski, Lindo Patterson, Cecile Pickart, Sidney Rieder, John Richard, James Robinson, Zelda Rose, Keith Schray, Steve Seeholzer, Frank Solomon, Michael Summers, Jessie Warms, Mary Wimmer, Keith Wilkinson, James Willard, Harland Wood and Alvin Zipursky.
This autobiography/biography was written at the time of the award and later published in the book series Les Prix Nobel/ Nobel Lectures/The Nobel Prizes. The information is sometimes updated with an addendum submitted by the Laureate.
Irwin Rose died on 2 June 2015.
Nobel Prizes and laureates
Six prizes were awarded for achievements that have conferred the greatest benefit to humankind. The 12 laureates' work and discoveries range from proteins' structures and machine learning to fighting for a world free of nuclear weapons.
See them all presented here.

