Advanced information
Advanced information:
The discoveries of human papilloma viruses that cause cervical cancer and of human immunodeficiency virus (pdf)
The discoveries of human papilloma viruses that cause cervical cancer and of human immunodeficiency virus
Professor Harald zur Hausen, emeritus Scientific Director and Chairman of the Management Board of the German Cancer Research Center in Heidelberg, has made seminal observations that identify novel human papilloma viruses as key contributors to cervical cancer. Cervical cancer is the second most common cancer among women. Professor zur Hausen’s discoveries include detection of novel human papilloma virus types, isolation of the human papilloma virus types 16 and 18 genomes, and expression of specific papilloma virus DNA genes integrated into the tumour host cell genome. These findings have led to an understanding of cervical carcinogenesis, a characterization of the natural history of the human papilloma virus infection, and paved the way for the development of preventive vaccines.
Professor Francoise Barré-Sinoussi, director of the “Regulation of Retroviral Infections” Unit, Virology department at the Institut Pasteur, Paris, and Professor Luc Montagnier, President of the World Foundation for Aids Research and Prevention, Paris, discovered human immunodeficiency virus-1 (HIV-1), the first human lentivirus. They characterized the virus based on its morphological, biochemical and immunological properties and demonstrated the capacity to induce massive virus replication and cell damage to lymphocytes. The initial discovery of Barré-Sinoussi and Montagnier was a basis for subsequent identification of this virus as the aetiological agent of acquired human immunodeficiency syndrome (AIDS). The discovery has led to epidemiologic surveys, tracing of the origin of HIV-1, identification of novel steps in the retroviral replicative cycle and generation of therapeutic as well as prophylactic options.
The search for an aetiological agent for cervical cancer
Carcinomas of the anogenital tract – particularly cervical cancer – had long been thought to be caused by a sexually transmitted agent (1). A prevailing hypothesis postulated herpes simplex type 2 virus (HSV-2) in the tumour aetiology, based on sero-epidemiological data and this was supported by nucleic acid hybridizations and cervical cancer induction in animals (2,3). However, Harald zur Hausen consistently failed to find HSV-2 DNA in cervical cancer cells when applying the in situ hybridization technique, although he had successfully used this method for identification of Epstein-Bar virus in transformed lymphoblastoid cells, Burkitt´s lymphoma and nasopharyngeal carcinomas (4-6). He postulated a role of another virus, human papilloma virus (HPV), in cervical cancer (7,8). He thought that human tumour cells, if transformed by a virus, would harbour viral genetic information integrated into the host genome. His “causality criteria” had added to the classical Koch’s postulates the criterion that the viral genome should be persistently present and transcriptionally active in the cancer. He suggested that the viral DNA could exist in a non-productive state in the tumours, i.e. not engaged in viral DNA replication or production of virus particles, and thus only be detectable by specifically searching for viral DNA. In 1976 he stated that “The condyloma (genital wart) agent has been entirely neglected thus far in all epidemiological and serological studies relating not only to cervical and penile, but also to vulvar and perianal carcinoma. This is particularly unusual in view of the localization of genital warts, their mode of venereal transmission, the number of reports on malignant transition, and the presence of an agent belonging to a well characterized group of oncogenic DNA viruses”(8). It had been long known among dermatologists that inoculation of extracts from condylomata acuminata and laryngeal papilloma could cause skin warts and occasional malignant conversion of genital warts into squamous cell carcinomas, usually at vulval and penile sites (9-12).
The awarded discovery of human papilloma virus causing cervical cancer
In 1974 zur Hausen published his first report at attempting to find HPV DNA in cervical cancer and genital wart biopsies by hybridizing tumour DNA with cRNA obtained from purified plantar wart HPV DNA (5). HPV particles had been detected in plantar warts by electron microscopy, and plantar cRNA was used as a probe since HPV could not be propagated in vitro (10,11). Hybridization was subsequently achieved in biopsies from such plantar warts and from verrucae vulgaris (skin warts), while biopsies from condyloma acuminata and cervix cancer were consistently negative. Since neither tumour type scored positive despite evidence of typical HPV particles in the condylomata lesions, he suspected genetic heterogeneity among HPV virus types (13). Harald zur Hausen´s theory of HPV aetiology for cervical cancer was supported by later work by Meisels and Fortin (14), who had described koilocytotic atypical cells as the manifestation of papilloma virus-induced cytopathic change in cervical dysplasia. The subsequent identification of papilloma virus-like particles in such cells by electron microscopy further supported the idea (15). Harald zur Hausen’s team subsequently used DNA purified from virus particles of different plantar warts to generate probes that allowed detection of distinct restriction enzyme cleavage patterns in HPV isolates from many patients (Fig. 1) (16-25). This led to the identification of multiple HPV strains 1-3 (16). Another isolate, HPV4 described a year later was clearly different from the first three isolates as it cross hybridized neither to HPV 1-3 nor to DNA from condylomata acuminata or larynx papilloma, further exemplifying the heterogeneity among HPV types (16).
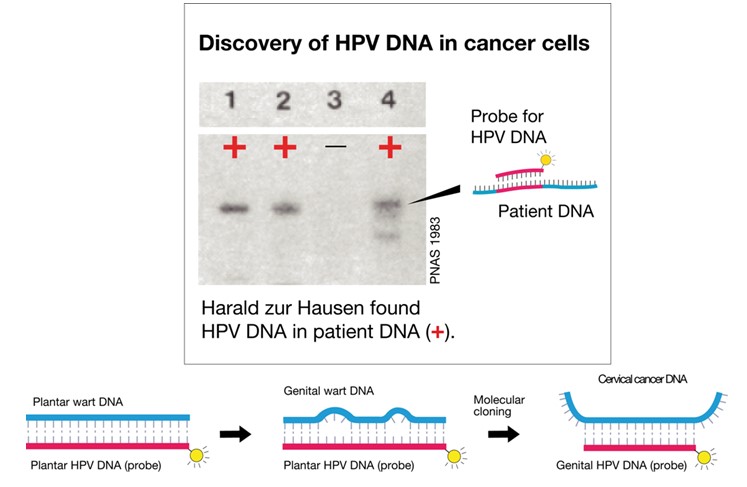
Harald zur Hausen continued to apply nucleic acid hybridisation at low stringency conditions to screen for novel HPV types, and developed a cloning procedure that led to the identification of the first genital HPV type, HPV6 (26). DNA from this type was repeatedly isolated from genital warts and condylomata acuminata (26,27) and the virus is today known as the major cause of condylomata. Subsequently, zur Hausen identified the closely related HPV11 from laryngeal papilloma and from genital warts (28). However, this still did not link HPV to cervical cancer. Later, a few malignant tumours, including two samples from invasive cervix carcinoma, showed weak but persistent cross hybridization with HPV11 DNA (29,30). This DNA was cloned and designated HPV16 DNA (31). Subsequently, zur Hausen demonstrated that more than 50% of cervix cancer biopsies scored positive for HPV16 DNA (Fig. 2); in addition, cross-hybridizing vulvar or penile cancers and condylomata acuminata specimens were found. Negative samples from African patients with cervical cancer led to an extensive search for additional HPV types (32). Again, combining low stringency hybridization with a mixture of HPV9,10 and 11 DNA probes resulted in the cloning of HPV18 DNA, while controls from benign tumours, normal cervical tissue and genital warts remained negative (33). It became clear that the typical precursor lesions of anogenital cancer, cervical intraepithelial neoplasias and Bowen’s disease also frequently contained the HPV16 or 18 types (32). Harald zur Hausen’s research team had now identified the two HPV types most frequently found in cervical cancer: HPV16 and HPV18 DNA are present in 82% of the patients with invasive cervical cancer. DNA from these types was also identified in cell lines derived from various cancers of the cervix (34-38).
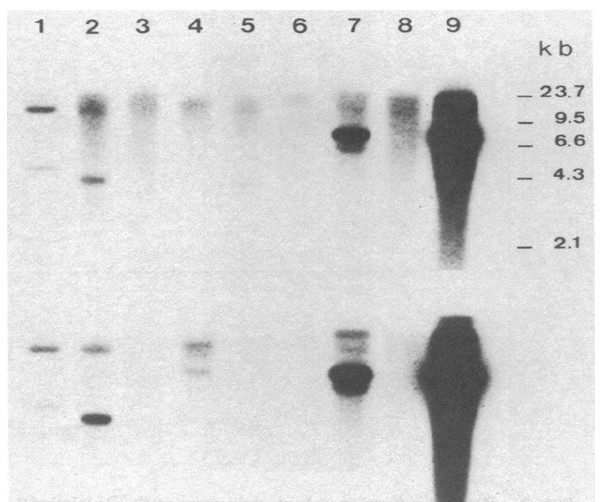
His most important findings were the identification and molecular cloning of the HPV16 and HPV18 genomes, and the associated demonstration that a majority of cervical cancers contained DNA that hybridized under stringent conditions to probes from HPV16 or HPV18 (31-36). His observations demonstrated that; papillomaviruses were etiologically involved in this cancer and infection by more than one HPV type could result in cervical cancer. Finally he demonstrated that parts of the HPV DNA was integrated into the host genome in cervical cancer cell lines including the HPV16 and 18 viral E6 and E7 genes which were preferentially retained and expressed in the tumors (47). These results provided an opening to study the role of the transfered viral DNA genes in malignant transformation. It showed that viral gene expression of the E6 and E7 genes were required for maintenance of the tumorigenic phenotype and for maintaining the transformed phenotype. These discoveries by Harald zur Hausen led to a paradigm shift in the field. Much new information has been learned through the efforts of many scientists. This is briefly summarized below to indicate the further and continued importance of the discovery.
Human papilloma viruses
The human papilloma viruses are small, non-enveloped, icosahedral DNA viruses that have a diameter of 52–55 nm. The viral particles contain a single double-stranded DNA molecule of about 8,000 base-pairs contained in a capsid composed of 72 pentameric capsomer proteins. The capsid contains two structural proteins – the late L1 and L2 – which are both virally encoded and expressed late in the replication cycle. The genomes of all HPV types contain approximately eight open reading frames (ORFs) that are transcribed from only one DNA strand. The ORF´s are classified into three functional parts: the early (E) region that encodes proteins (E1–E7) necessary for viral replication, the late (L) region that encodes the structural proteins (L1–L2) required for virion assembly, and a largely non-coding part that is referred to as the long control region (LCR) which contains cis elements necessary for viral replication and transcription. Today, we know of more than 100 HPV genotypes of which 40 infect the genital tract, and about 15 of these put women at high risk for cervical cancer. In the vast majority of cases, however, the immune system clears HPV infections before they cause harm (39).
The discovery led to understanding of the HPV pathogenesis
Infection with the HPV16, 18, 31 and 45 types are considered to give a high risk for development of cervical cancer, while HPV6, 11, 42, 43 and 44 are low risk, non-oncogenic types linked to genital warts and non-malignant lesions (39). HPV infects keratinocytes of the basal cell layer, usually in micro lesions of skin or mucosa. The population of virus-infected cells spreads laterally as they divide, and some cells migrate into the supra basal differentiating cell layers, where viral genes are activated, viral DNA replicates and capsid proteins are formed. Virus particles are released, thus spreading the population of infected cells (Fig. 3) (40). Harald zur Hausen demonstrated that tissue from cervical dysplasia as well as from premalignant bowenoid lesions contains episomal HPV-DNA, and prospective studies showed that approximately 90% of the HPV16 and 18 positive tissues became negative within two years (Fig. 3 and 4) (32, 36, 41,42). Less than 10% of infected individuals develop dysplasia and most of these show a mild form that either regresses or that fails to progress (39). Fifty to 80% of a sexually active population becomes HPV infected during a lifetime (39,40,43). The actual lifelong risk for cervix cancer in those infected is not known and depends on type of HPV infection plus the genetic and immunological background (41,42). Women with a persistent “high-risk” HPV infection (i.e. infected with an oncogenic HPV type) are 300 times more likely to develop high-grade neoplasia (41). Often, such patients lack an effective cell mediated immunity thought to be important for eliminating HPV infected cells. Organ transplant recipients and HIV infected patients also have a more severe and recalcitrant disease, higher viral loads, infections with unusual HPV genotypes and greater propensity for HPV related malignancies, especially cervical cancer among women and anal cancer among men (Fig. 5) (44,45).
Identification of factors that affect HPV induced malignancy
The majority of cervical cancers develop from squamous cells. Cervical cancer is characterized by a well-defined pre-malignant dysplasia that can be diagnosed by pap smears (Papanicolaou test) i.e. by cytological examination of sampled cervical cells and confirmed by histological analysis of cervical material. Such pre-malignant changes represent a spectrum of histological abnormalities ranging from CIN1 (mild dysplasia) and CIN2 (moderate dysplasia) to CIN3 (severe dysplasia/carcinoma in situ). Cytological and histological examinations are however insufficient to identify, at this early stage, the few women with abnormal smears that will develop invasive disease (39-42).
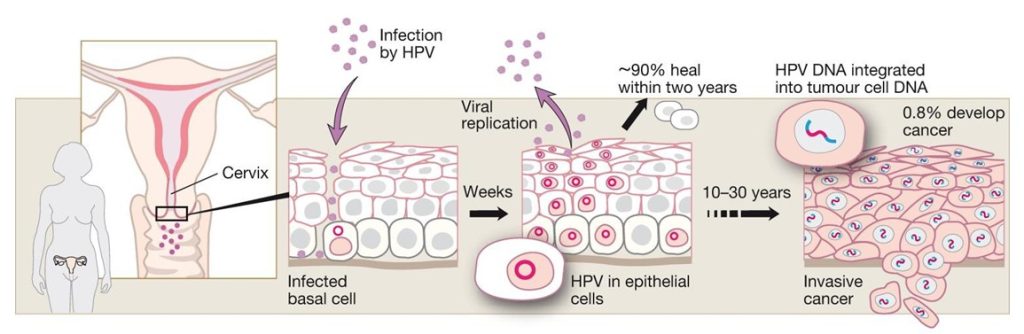
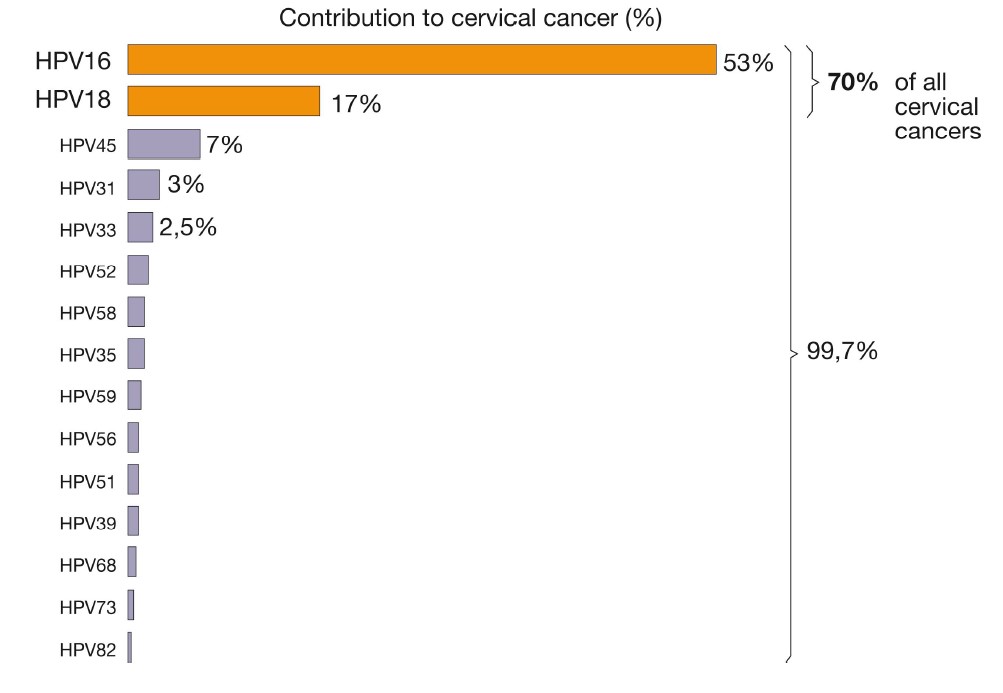
Studies of 3,000 women from 25 countries found HPV16 and 18 in >70% of the malignant cases, with only minor geographical differences (39,43). The next five most prevalent types accounted for another 20%, whereas the remaining 10% are caused by a wide variety of HPV types (Fig.6) (43,46). Sensitive PCR assays have since improved the detection of viral DNA, and accumulated epidemiological data have confirmed zur Hausen’s original discoveries. Indeed, as much as 99.7% of the cervical cancers studied have been found HPV-positive in some reports (46), which provides compelling evidence for the HPV aetiology of the disease (fig 5). Cervical cancer tissues harboured the viral DNA integrated into the host-cell genome (Fig. 4) (47), with viral sequences encoding the early genes E6 and E7; the disruption of their control region results in a loss of negative-feedback control of the oncogene expression (Fig. 3). Whereas the prevalence of integrated forms of HPV increased as the disease progressed, integration itself was followed by a decrease in viral load. However, integration was found at a single chromosomal site in the tumour cells of almost all cervix cancers examined, consistent with the idea that cervical cancer is a clonal disease.
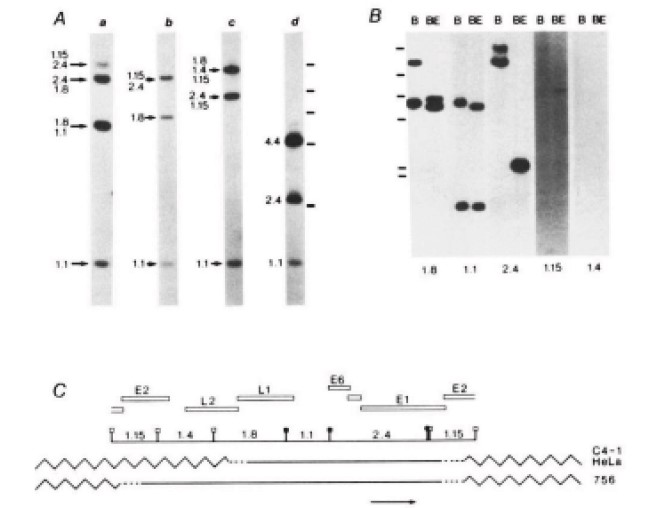
The E6 and E7 genes of the virus types found in cervical cancer were expressed from viral DNA integrated in the tumour cell genome (Fig. 4) (38,47). This was observed in primary biopsy specimens from these cancers and in most cell lines derived from the cancers. Subsequent in vitro experiments showed that the two oncogenes of high risk, anogenital HPV modified a large set of cellular genes, by changing their expression pattern by functionally inactivating them or by targeting those for degradation (48,49). Thus, integration of HPV-DNA into the cellular DNA is considered to play a central role in the induction and maintenance of the transformed cells.
When transfected, the E6 and E7 genes of the high risk HPV types immortalize human keratinocytes, in contrast to low risk strains (50). The E6 protein binds p53 and abrogates its tumour suppressive and transcriptional properties (51) by promoting ubiquitination of p53 and its subsequent proteolysis through interaction with the E6AP ubiquitin-protein ligase (52). The E7 protein interacts withthe retinoblastoma susceptibility gene product pRb and related proteins (53). As a consequence, it releases the transcriptional activator E2F from a complex with Rb, thereby allowing E2F to activate genes engaged in cell cycle progression. E6 also targets other proteins including the focal adhesion protein paxillin (54), the interferon regulatory factor 3 thus blocking viral induction of IFN-b, and renders the malignant cells non-responsive to TNF-a mediated changes in the AP-1 complex (55,56).
The above illustrates the multifunctionality of many viral oncoproteins,: they impair a multitude of cellular functions that aim to suppress tumour cell functions such as DNA repair and programmed cells death, a property that eventually leads to progression of the carcinogenic state and malign tumours. Thus, mutations causing chromosomal alterations, loss of heterozygosity, and proto-oncogene and telomerase activation are thought to have important roles in the virus-induced cervical carcinogenesis (57-60).
The discovery led to new hope for cancer prevention
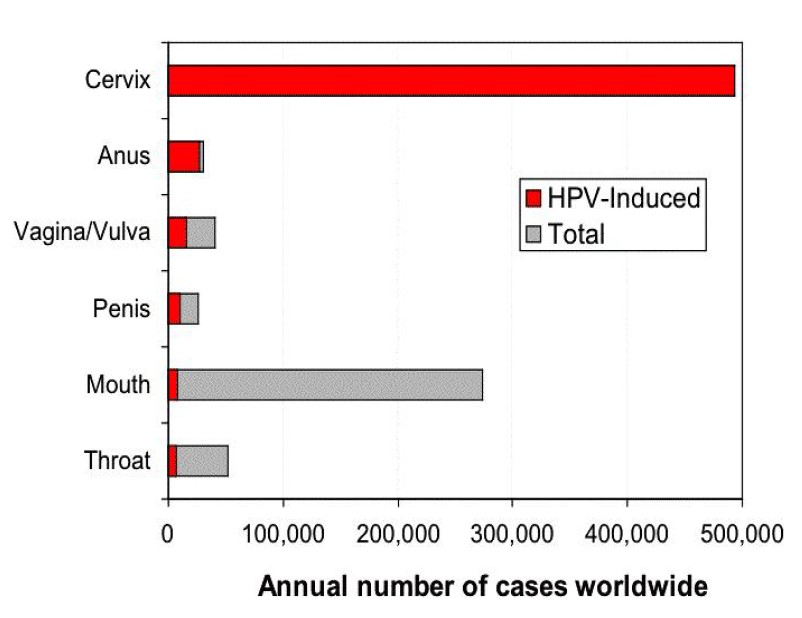
Cervical cancer is the second most common life-threatening cancer among women. About 500,000 new cases of cervical cancer are diagnosed every year with a 10-fold higher incidence in developing countries, resulting in 250,000 deaths (39,61,62). It affects about 1 per 123 women and kills about 9 per 100,000 per year. Increased development of cervical carcinoma has been associated with; 1) early age of sexual debut; 2) multiple sexual partners; 3) poor sexual hygiene; 4) poverty; 5) immunosuppressed state of the host; 6) carcinogenic co-factors; 7) genetic background and 8) epidemiologic data (39-41). Harald zur Hausen’s discovery of a restricted number of HPV types as the aetiological cause of cervical carcinoma led to a hope for prophylactic vaccines. Experiments made in dogs and rabbits using purified papilloma virus structural proteins – that spontaneously assembled into virus like particles (VLPs) – resulted in effective protection against the primary infection. This provided the background for further development of vaccines against human high-risk HPV types based on the synthesis and self-assembly of the major virus capsid protein, L1. Studies by Schiller and Lowy in the early 1990s showed that expression of the HPV’s L1 protein in baculovirus or yeast led to the self-assembly of VLPs (63). These empty shells of L1 containing capsids mimicked HPV’s shape, and neutralizing anti-VLP antibody responses in VLP-immunized women were up to 10 fold greater than those identified in natural infections (64). A quadrivalent vaccine available today includes immunogens for HPV types 6, 11, 16 and 18, and has been shown to be effective at protecting against HPV6 and 11 condylomas as well as HPV16- and 18-related cervical intraepithelial neoplasia (CIN) stage 2/3 (high grade cervical intraepithelial neoplasia, a precursor of invasive cervical cancer) in HPV naïve women. The vaccine has also prevented anogenital condylomata and vaginal/vulval dysplasias in HPV naïve volunteers. A divalent HPV VLP vaccine has also been developed and includes L1 proteins from HPV types 16 and 18. Both vaccines have been licensed, appear to have few side effects, and have generated >90% protection in phase-III trials (65,66). These two HPV vaccines potentially provide protection against the two types of HPV that cause approximately 70% of invasive cervical cancers worldwide (Fig. 6) (67). However, discoveries whether these vaccines are effective in preventing not only against cervical lesions but also cervical cancer and death must await the collection of epidemiological data during the coming decades (68). In addition, the duration of the vaccine’s protection is unclear: do they provide life-long immunity or will booster doses be needed? Since the vaccines aim to protect against the two virus strains that cause 70% of the cervical cancers, will other strains emerge as a major cause of the tumours?
The discovery of human immunodeficiency virus (HIV)
A new pandemic of acquired immune impairment
In 1981 a new serious medical syndrome was described in California and New York (69). The report identified clusters of previously healthy young men who suffered from different life threatening medical conditions previously not seen in this population. A task force led by the Centers for Disease Control (CDC) was set up, and defined the disease as the “Acquired Immune Deficiency Syndrome” (AIDS) (70). It established that the syndrome was new and that the number of cases increased rapidly. A huge epidemiological survey initiated by CDC in 1982 concluded that the AIDS syndrome had spread globally. A subset of the population at particular risk for this syndrome appeared to be homosexual males and intravenous drug users, but there were also some cases in heterosexuals, haemophiliacs and immigrants from Haiti. The immunodeficiency was associated with rapid elimination of CD4+ T cells and antigen presenting cells (71). The clinical AIDS spectrum was defined as repeated opportunistic infections, specific malignancies and autoimmune phenomena occurring in previously healthy adults with no history of inherited disorders. Patients acquired specific tumours and opportunistic infections with pneumocystis jiroveci, Candida species, Cryptococcus, Aspergillus, Toxoplasma, Mycobacterium avium, Herpes virus, Varicella or Cytomegalovirus (72-74). Mycobacterium tuberculosis infection was a predominant cause of death together with Pneumocystis jiroveci pneumonia. Malignancies associated with AIDS included an aggressive type of Kaposi’s sarcoma caused by human Herpes virus 8, EBV-associated lymphoma, HPV-induced cervical cancer, and Hodgkin’s disease (75). The disorder also manifested as slim disease due to chronic incurable diarrhoea, particularly in Africa. T lymphocyte hypo-responsiveness and imbalance of T helper and suppressor cells was a hallmark and appeared to predispose to the opportunistic infections and the spectrum of malignancies (71). Epidemiological studies had already been established that AIDS was transmitted sexually, via placenta to foetuses and via transfusion by plasma and coagulation products (76). However, it was initially not obvious that AIDS was one disease entity and that the symptoms which involved almost all organ systems could have one single cause. Nevertheless at the end of 1982 several virus laboratories attempted to find a cause for the AIDS syndrome.
The awarded discovery of lymphadenopathy associated virus (LAV)/human immunodeficiency virus (HIV)
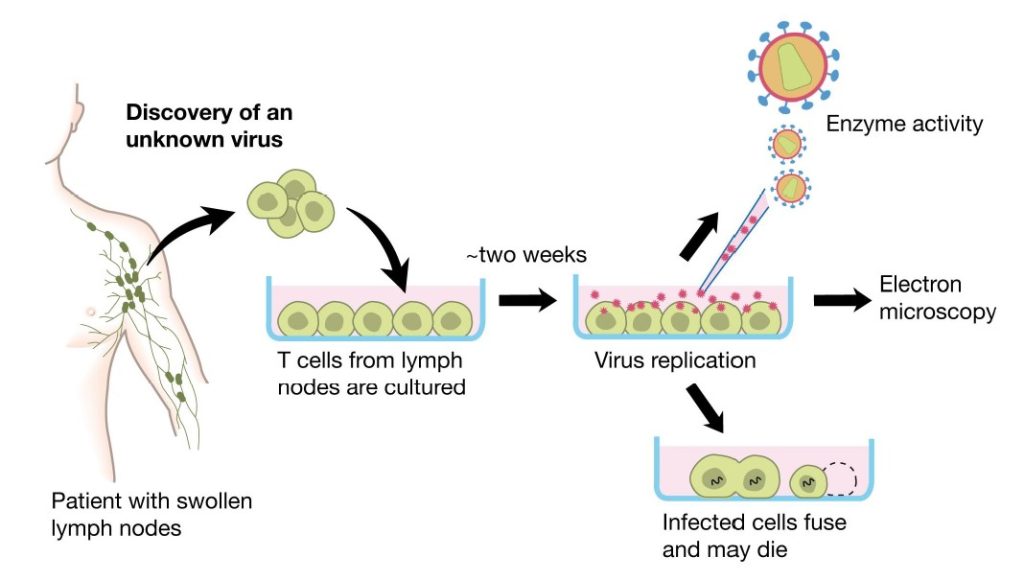
A number of pieces of evidence pointed towards a retroviral origin for the acquired immune deficiency; the clusters of patients affected, the transmission via filtered blood products and the establishment of loss of CD4 T helper lymphocytes. A working group led by Francoise Barré-Sinoussi and Luc Montagnier at the Viral Oncology Unit at Institut Pasteur, Paris, collaborated with clinicians in order to explore this hypothesis. In 1983 Barré-Sinoussi and Montagnier isolated lymph node cells from patients with lymphadenopathy and early signs of acquired immune defect. Barré-Sinoussi and Montagnier therefore decided to regularly check for virus by analysing lymphocytes from patients at the early stage of infection that should still have CD4+ T cells. Furthermore, it was clear that such individuals often had a generalized lymphadenopathy that preceded AIDS, which potentially would allow detection of the virus at this site. Thereby starts the process that led to the discovery awarded the Nobel Prize.
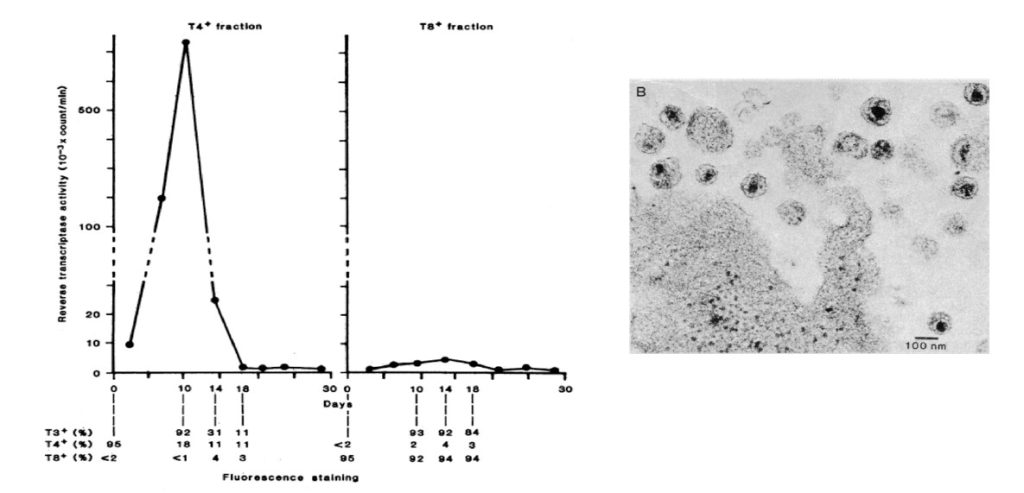
They cultured purified lymphocytes from such patients in vitro in the presence of the phytohaemagglutinin (PHA)-mitogen, interleukin-2 (IL-2) and anti-interferon-a in order to allow T cell proliferation in the search for a potential viral aetiology. The profound depletion of CD4+ T lymphocytes in affected individuals indicated that these cells could be targets and destroyed as a consequence of the infection. Supernatants from cultured lymphocytes were shown to contain reverse transcriptase (RT) activity (77,78), using the Mg2+ and polyadenylate-oligodeoxythymidylate conditions also described for HTLV-I (79,80). The RT activity was not found in the presence of Mn2+, indicating that the activity was caused by a mammalian retrovirus. Detection of the RT activity was dependent on continuous addition of fresh mononuclear cells from normal blood donors, hinting that there was virus-induced cell death in the cultures (Fig. 7). Electron microscopy subsequently identified typical 90-130 nm large retroviral particles budding from cell membranes (Fig. 7). The virus particles had a density of 1.16 in sucrose gradients, characteristic of most retroviruses. Cell free supernatants from these cultures transferred the virus to umbilical cord blood and to mononuclear cells from healthy donors. Antibodies to the virus were identified in sera from patients by immunofluorescence analysis. Patient antisera specifically precipitated a 25-kDa protein (now called p24Gag) from radio-labelled virus. The protein did not react with type-specific antisera to the HTLV-1 p19 and p24 proteins, suggesting a new, previously unidentified retrovirus. The clinical isolate had, unlike HTLV, no transforming activity on T lymphocytes. Barré-Sinoussi and Montagnier concluded that they had discovered a new human, non-transforming retrovirus containing a major p25 protein, similar to that of HTLV-I but with different antigenic properties. Cultured cells from patients generated extracellular release of viral particles that could infect lymphocytes from healthy adults and newborns. This virus isolate was called lymphadenopathy associated virus (LAV) in 1983 (Fig. 7) (81).
Barré-Sinoussi and Montagnier also studied a pair of siblings with haemophilia B (treated with factor VIII). One brother was healthy but the other had symptoms of AIDS (Kaposi’s sarcoma). A virus similar to LAV was isolated in both cases with a typical lentivirus D-type of morphology, with cylindrical conical core (Fig. 7) distinct from the large spherical cores of HTLV-I and HTLV-II (78). They called this virus immunodeficiency associated virus (IDAV-1 and IDAV-2, respectively). The IDAV-1 and IDAV-2 had a core p25 protein identical to that of LAV as assessed by specific antisera. They also isolated virus from peripheral blood obtained from diseased patients and demonstrated that this virus had a cytopathic effect that could promote giant cell formation in cultured infected T lymphocytes (82).
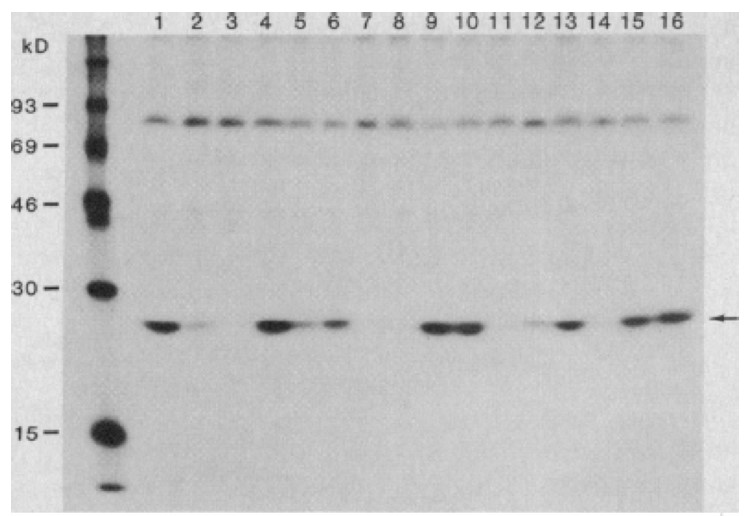
Furthermore, antibodies to their new virus were detected in individuals at risk for AIDS as well as those with AIDS and AIDS-related conditions but not in patients with other diseases, supporting the concept that LAV/IDAV was crucial to the disease (Fig.8) (83-85). In subsequent studies Barré-Sinoussi and Montagnier isolated the same type of virus from sexually infected individuals, haemophiliacs and blood transfused patients; evidence was also found for mother to child transmission (84-88).
Thus, within one year Barré-Sinoussi´s and Montagnier´s group had characterized a virus from pre-AIDS and AIDS cases that had the following unique properties: a 90-130 nm sized retrovirus with cylindrical core (D-type) of lentivirus type that targeted CD4+ T cells. It showed cytopathic effects in T cells plus an extensive virus replication that led to cell free virus transmission. It did not cause cell transformation and had a unique p24 protein and his clearly separated the LAV/IDAV virus from HTLV-I and –II (89-92). The discovery was accepted by the research community and resulted in an explosion of scientific breakthroughs as illustrated below.
Contributions of more lentivirus clinical isolates by other research groups and defining the role for the virus in the acquired immunodeficiency syndrome, (AIDS)
Professor Robert Gallo’s group at NIH described the detection of a novel HTLV-like virus from a vast number of patients with AIDS or pre-AIDS in 1984 (93). The virus shared some properties with HTLV-1 and HTLV-2 and was denoted HTLV-III; however, it showed considerable similarities with LAV-1. By using newly developed immunological reagents and techniques, antibodies against HTLV-III proteins were found in a majority of AIDS and pre-AIDS cases and in clusters of homosexual men (94-96). Professor Jay Levy’s group in San Francisco also identified a D-type retrovirus, of the lentivirus group, which was structurally related to LAV-1 and HTLV-III, from AIDS patients and patients with lymphadenopathy. The virus was denoted AIDS-associated retroviruses (ARV) (97). The American and French teams agreed later that LAV-1/IADV-1/HTLV-III and ARV was the same type of virus (Fig 9). An international virus taxonomy consortium chose in 1985 to name the virus Human Immunodeficiency Virus type 1, or HIV-1 (108-110).
The virus belongs to the lentivirus subfamily of retroviruses which is distinct from the previously identified human retroviruses, the oncogenic leukaemia viruses HTLV-I and II. Lentiviruses group contain viruses that cause persistent viremias in monkeys, cats, horses, cattle and sheep. Extensive seroepidemiological work by several research groups led by Robert Gallo, Luc Montagnier, Max Essex, William Haseltine, Jay Levy and Robin Weiss further confirmed the causative role of the virus in the development of acquired immunodeficiency syndrome, AIDS (98-103).
Detection of the origin of HIV and understanding the epidemiology of AIDS
Viral gene trees can deliver powerful insights into ecological and evolutionary processes. Such phylogenetic analysis of HIV-1 has traced its origin to the simian retrovirus SIVcpz, which has been found in chimpanzee colonies in southern Cameroon (Fig 10) (111). It is thought that SIVcpz was passed to human hunters from captured chimpanzees, and that the animal virus during its replication in humans underwent many genetic changes to evolve into a human, highly transmissible pathogen that causes AIDS. The origin of the current HIV pandemic has been a subject of great interest and speculation. Viral archaeology sheds light on the geography and timescale of the early diversification of HIV-1 in humans. A lymph node biopsy taken in 1960 from an adult female in Kinshasa, have been compared for HIV gene sequences with a virus characterized from a plasma sample from a nearby village from 1959 and made it possible to performe the first evolutionary analysis of pre-AIDS ‘fossil’ HIV-1 sequences. The analysis supports the idea that diversification of HIV-1 in west-central Africa occurred long before the recognized AIDS pandemic (112). It estimates that the date of that common ancestor is between 1902 and 1921. Independent transmission events occurred between 1902 and 1941 (112, 113), which spawned three HIV-1 groups: the major (M), the outlier (O) and the nonmajor outlier (N) categories.
Strains related to the M and N groups have been found in chimpanzees; however, recent evidence suggests that group O HIV-1 may have originated in gorillas (114). The virus is believed to have spread among humans along the Congo River into Zaire (Fig. 10).
The earliest documented human case of HIV-1 infection, with the M strain, has been traced to a blood sample from 1959 (115).
The three groups of HIV-1 have major genetic differences. Most HIV-1 infections are caused by group M viruses, and these are divided into 9 subtypes known as clades (A–D, F–H, J, and K). The viral genomic sequences can differ by 15%–20% in distinct clades. The most common clade in the Americas, Europe, and Australia is clade B, whereas clade C predominates in the most severely affected part of the world, southern Africa. The cumulative number of individuals infected with HIV-1 since the pandemic began exceeds 60 million people, and deaths due to AIDS have been estimated to 25 million. At the end of 2007, the Joint United Nations Programme on HIV/AIDS (UNAIDS) and the WHO calculated that there were 33.2 million people living with HIV-1, that 2.5 million individuals were newly infected with HIV-1 in 2007, and that 2.1 million people died of AIDS in that year (Fig. 11). There has also been a strong increase in the number of women infected with HIV-1 through heterosexual contact.
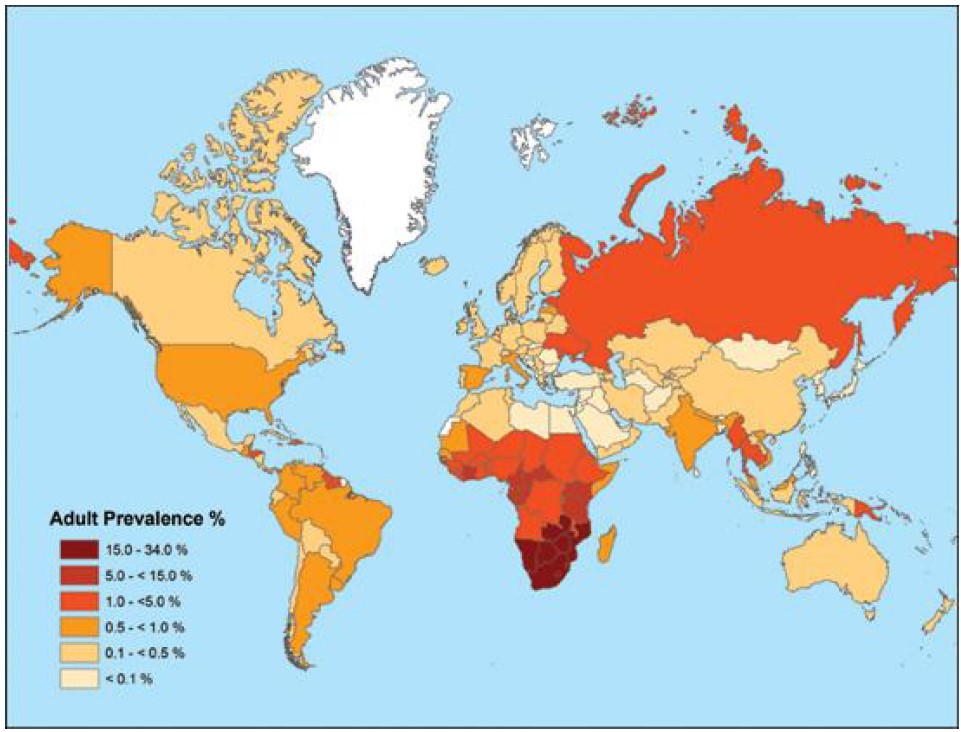
33.2 million individuals currently living with HIV-1 according to UNAIDS (134).
As of 2007, approximately 61% of individuals infected with HIV-1 in Africa were women. Almost 90% of the children infected with HIV-1 live in Africa. Overall, the global prevalence of HIV-1 is reaching a 1%, a level that strongly suggests a continued, high prevalence of AIDS in the decades to come (117).
Unravelling HIV-induced pathogenesis
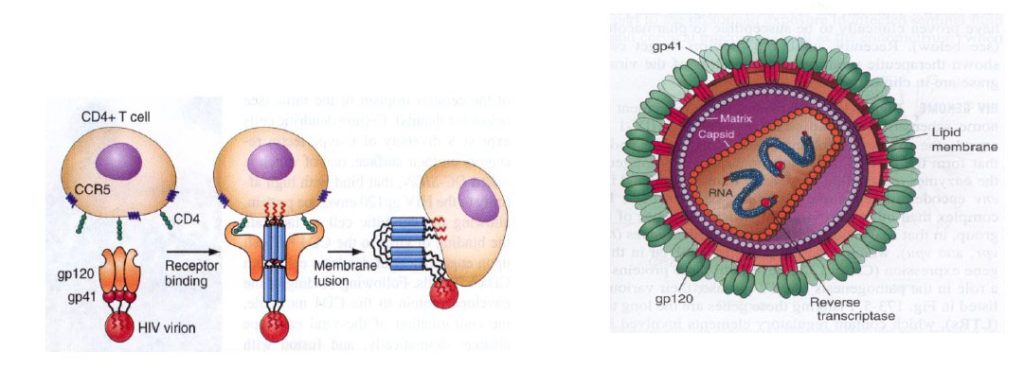
Both free and cell-associated HIV-1 can establish a mucosal infection. In the first hours of infection, virus and/or infected cells cross the cervicovaginal/penile foreskin mucosal barrier (Fig. 12). Infection starts by the gp120 envelope binding in trimeric form to the CD4 molecule, which results in a conformational change in gp120 that facilitates binding to a co-receptor and that causes the virus to fuse with the cell (118). The molecular architecture of native HIV-1 gp120 has been crystallized (118). There are two major co-receptors, CCR5 and CXC4 are primary receptors for chemokines. Certain dendritic cells express C-type lectin receptors that also bind the gp120 envelope protein with high affinity without resulting in viral replication. Productive infection mainly occurs in “resting” CD4+CD45+RO+T cells which can support viral replication because they are the most numerous target cells in the lamina propria (119). After proviral integration, virus production can occur in susceptible CD4+ T cells, macrophages and dendritic cells in the lamina propria (119). Expansion of infection from small founder populations of infected cells in the lamina propria subsequently spreads virus and infected cells, first to the draining lymph nodes and later systemically, to maintain virus production especially in the lamina propria of the gut associated lymphatic tissue (Figure 12) (120). Within six days after virus exposure, HIV overcomes the body’s initial defenses, ultimately resulting in an exponential viral burst. Immune responses are activated, but neutralizing HIV antibodies do not occur before 4 months after primary infection and insufficient to eradicate infection. Immune activation and inflammation supplies additional activated CD4+ T cells, which both sustain infection and elicit an immunosuppressive response that blunts host defences. Although increasing numbers of cytotoxic T lymphocytes (CTLs) partially control infection they do not prevent, in the absence of therapy, the slow and continued depletion of CD4+ T cells that is responsible for the occurrence of the immune deficiency that eventually leads to AIDS. Without treatment, about 9 out of every 10 persons with HIV will progress to AIDS within 10-15 years.
HIV-1 has developed a number of mechanisms to elude immune control. The most prominent of these is the extensive glycosylation of the external envelope glycoprotein, which protects neutralization epitopes. Furthermore, the viral targeting of the CD4 molecule results in CD4 helper T cell death, and HIV integration into the host-cell genome, which indicates that surviving cells are infected permanently. Another important factor is that the viral RNA replication cycle lacks normal proof-reading machinery; the viral genome therefore mutates at high rate to escape the host immune system (117).
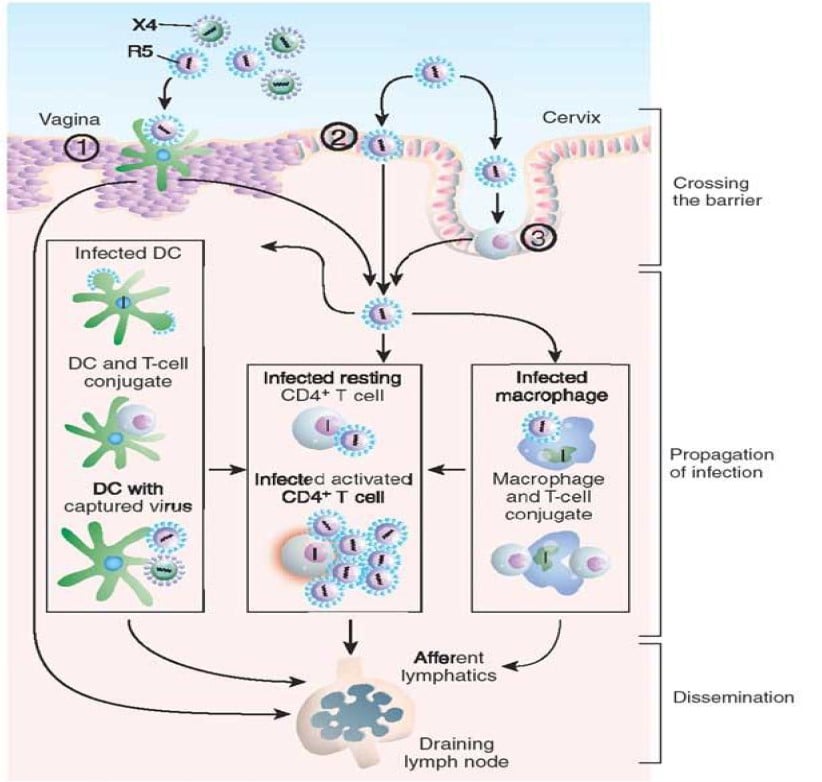
Understanding mechanisms for host defence in HIV-1 infection
During acute HIV infection in adults, a peak of ~107 HIV RNAs/ml plasma is followed by a marked decline over a few weeks to around 30,000 RNAs/ml concomitantly with the appearance of HIV-specific CD8+ cytotoxic T lymphocyte (CTL) responses suggesting that CTLs may be responsible for reducing virus levels (121-127). Furthermore, blocking these cells by administration of CD8-specific monoclonal antibodies prohibits the immune mediated reduction in viral load. Persistent control of HIV-1 infection is associated with the presence of CD8+ T cells that are capable of producing not only IFNg following contact with the cognate-peptide–MHC complex, but also other cytokines, such as IL-2 and chemokines, including RANTES and MIP-1a which are capable of blocking viral entry through occupancy of the HIV co-receptor CCR5. HIV-specific CD8+ T cells also release granzyme and perforin, which are capable of mediating cytotoxicity. Typically, HIV-specific CD8+ T cells from infected individuals who have low viral loads are polyfunctional and can carry out most or all of these effector functions (126). Conversely, HIV-infected subjects with high viral loads often have HIV-specific CD8+ T cells that are capable of only some of the normal effector functions and, at the extreme of the spectrum, their dysfunctionality might be reflected by the expression of PD1 (programmed cell death 1), a marker of T cell exhaustion. Proliferative capacity of both CD4+ and CD8+ T cells are also associated with control. In addition, genetic regulation of CCL3L1 (MIP-1aP) and CCR5 have been demonstrated to be important because they regulate CTL activity and other viral entry dependent mechanisms (128). By binding to CCR5 and promoting its sequestration, CCL3L1 can block HIV replication.
An apparently persistent suppression of viral replication coupled to no drop in CD4+ T cells occurs in about one of 300 persons infected with HIV (elite controllers). It is striking that, in a recent whole-genome association study of key determinants for host control of HIV-1, the six most significant protective determinants were within the MHC region. It is evident that in HIV infection, HLA-B molecules have a stronger impact on viral setpoint than HLA-A and HLA-C. A gag-specific but not env-specific CD8+ T-cell response was protective against disease progression (128). Therefore, it is not just the number of Gag epitopes presented by each allele that is important, but also the ability of the Gag-specific CD8+ T-cell response to drive the selection of escape mutations into less fit virus that have an impact on viral replication (128). On the other side, uncontrolled HIV replication cause non HIV specific immune activation and chronic inflammation that are strongly associated with acquiring opportunistic cancers, chronic virus infections or Mycobacterium tuberculosis as well as end-organ damage, including cardiovascular, hepatic and renal dysfunction.
Development of antiretroviral drugs
The discovery of HIV allowed for a rapid dissection of the viral replication cycle (Fig 13) (129). Drug developers initially focused on finding inhibitors to the viral reverse transcriptase, and the nucleoside analogue, azidothymidine was already on the market by 1987. Further research produced a large number of different drugs targeting the HIV encoded reverse transcriptase (Fig 13). It rapidly became clear that the ability of HIV-1 to generate drug-resistant mutants meant that therapy would require a combination of agents affecting different proteins involved in viral replication (130, 131). In order to develop antiretroviral therapy (ART) subsequent development was focused on the protease enzyme that was successfully exploited for novel antiviral drugs. The protease cleaves viral pro-polypeptides into final proteins. The most recent developments include integrase inhibitors (2007) and viral entry inhibitors including CCR5 receptor antagonists (2008) as well as fusion inhibitors (128). The successful development of these compounds allows combination therapy that has dramatically increased the life expectancy of AIDS patients in developed countries (131, 132). The antiretroviral treatment provides several beneficial effects: reduction of the virus load with a concomitant reduction in virus spread, a cessation of T cell death, and therefore also the re-emergence of a functional immune system. The development of these compounds was highly dependent on the molecular cloning of the viral genome, which was a prerequisite for performing modern high throughput screening for new drug targets at research centres and within the pharmaceutical industries. The present antiretroviral drugs are suppressive, and they cannot cure infection with HIV-1. However, successful antiretroviral therapy results in life expectancies for persons with HIV infection now reaching similar levels to those of uninfected people. Currently, 3 million people are being treated with anti-retroviral drugs (133-135). Unfortunately this has not brought the epidemic under control and approximately 2.5 individuals become infected with HIV-1 for each new person started on ART (136). Furthermore, WHO has estimated that >6 million people would require treatment globally and set the ambitious target of providing treatment to additional 3 million people in resource-poor settings (135-137).
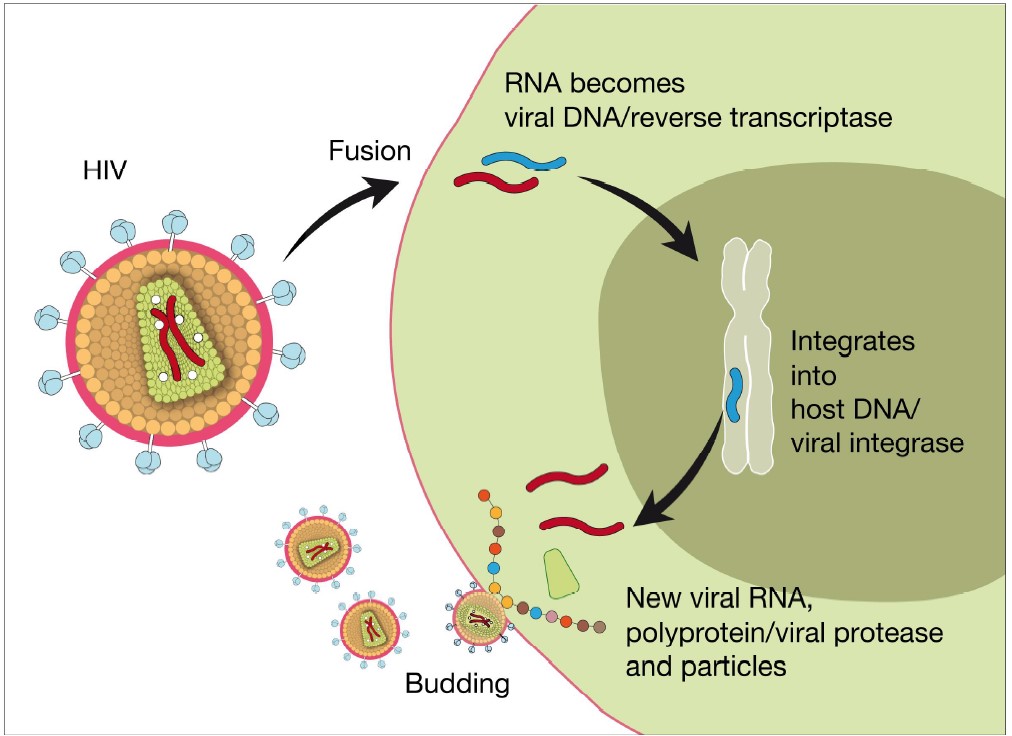
Investigating how protection from infection is maintained
Resistance to infection reflects some combination of genetic factors, innate resistance, and a probably very rare acquired immune control. Genetic studies have demonstrated that a deletion in the CCR5 gene (CCR5d32 deletion) provides substantial protection from HIV-1 infection (137). Members of the APOBEC (apolipoprotein B mRNA-editing catalytic polypeptide) family belong to cellular cytidine deaminases that represent a recently identified group of proteins induced by IFNa that protect from infection by retroviruses (139). Yet, HIV-1 is largely immune to the intrinsic antiviral effects of APOBEC proteins because it encodes a unique Vif gene (viral infectivity factor), an accessory protein that is critical for in vivo replication of HIV-1 due to VIF´s ability to inactivate APOBEC (140). Another intercellular antiviral factor, the tripartite motif–containing 5-α (TRIM5-α) gene, inhibits the ability of different retroviruses to infect human and monkey cells by blocking virus capsid uncoating (141). HIV-1 infection is restricted by the old world monkey TRIM5-a gene, but not by the human counterpart explaining why HIV can infect CD4+ T cells (142). Cellular miRNA can also interfere with HIV replication; however, HIV-1 can modify miRNA expression profiles in the infected cell (143). Several chemokines including, RANTES, MIP-1a/b and antimicrobial peptides such as a/b-defensins and LL 37 also inhibit HIV-1 replication locally. Thus, a number of innate mechanisms are of central importance for protection against HIV-1.
Furthermore, neutralizing IgA in the genital tract clearly contributes to protection against HIV-1. However, there are indications that these IgA antibodies are predominantly directed against host specific epitopes involved in virus binding and fusion, and it remains to be determined if these are natural or of acquired origin.
Medical attempts to induce protection include male circumcision, and randomized, controlled clinical trials have demonstrated the efficacy of this in reducing female-to-male transmission of HIV-1 by 50%–60% (144). To halt the present pandemic, a vaccine that protects from HIV-1 infection is urgently needed. An absolute worldwide commitment to this goal has been established; however, attempts to develop a protective vaccine have been severely compromised by our incomplete understanding of HIV-1 protective immunity. Furthermore, safe vaccine prototypes have so far failed to induce a sufficiently potent immune response against HIV-1 (145).
Conclusions
The global public health burden attributable to human papilloma viruses is considerable. Infection by the human papilloma virus is the most common sexually transmitted disease, afflicting 50-80% of the population. zur Hausen was first to recognize and demonstrate the association between cervical cancer and a subset of previously unknown genital human papilloma viruses. He unravelled the heterogeneity of the papilloma virus family, the genotype specificity in diseased individuals, the lack of productive viral replication, and the persistence of integrated viral DNA in tumour cells. This has led to an understanding of mechanisms for papilloma virus-induced carcinogenesis and the predisposing factors for viral persistence and cellular transformation. More than 5% of all cancers worldwide are caused by persistent infection with this virus. In addition to cervical cancer, other malignancies in both men and women such as oesophageal, oropharyngeal, penile and anal cancer are associated with this virus. Pooled data from case–control studies indicates that human papilloma virus can be detected in 99.7% of women with histologically confirmed cervical cancer. The discoveries by zur Hausen have laid the foundation for subsequent development of vaccines against the high-risk papilloma viruses that cause about 70% of cervical cancers. The vaccines may also prevent unnecessary surgical interventions aimed to treat precancerous lesions and may substantially reduce the global economic and social burden of cervical cancer.
The discovery of Barré-Sinoussi and Montagnier of a novel lentivirus made it possible to perform molecular cloning of HIV-1. This has allowed unravelling of important details of its replication cycle and how the virus interacts with its host. This led quickly to the development of diagnostic tools for identification of patients and screening of blood products, which has limited the spread of the pandemic. The unprecedented development of several classes of new antiviral drugs is also a result of elucidating the details of the viral replication cycle. The combination of prevention and treatment has resulted in a substantial decrease in disease spread and longer life expectancies in developed countries. Despite these advances, considerable problems persist. Firstly, global access to and use of effective interventions remain limited in the developing world. Secondly, progress in HIV-1 vaccine development is restricted by an incomplete understanding of immunity to HIV-1 infection and induction of potent protective immune responses. Lastly, we have no means to cure infection with HIV-1 due to the inherent difficulties in eliminating either the provirus in infected cells or the cells themselves, as AIDS patients carry a latent pool of cells with the potential to start producing viruses also after long dormant periods. Today, 27 years after identifying the AIDS syndrome and 25 years after HIV-1 was discovered, we have gained remarkable insight into this new pandemic.
Acknowledgements
A special thanks to Professor Björn Vennström for constructive critique, careful proof reading and supplementation of data.
Jan Andersson
Professor of Infectious Diseases
Department of Medicine, Karolinska Institutet, Karolinska University Hospital, Huddinge
References
1: Gagnon, F. Contribution to the Study of the Etiology and Prevention of Cancer of the Uterus. Am. J. Obstet. Gynecol., 1950; 60: 516-522.
2: Rawls, W. E., Tompkins, W. A. F., and Melnick, J. L. The Association of Herpesvirus Type 2 and Carcinoma of the Uterine Cervix. Am. J. Epidemiol.,1969: 89: 547-554.
3: Frenkel, N., Roizman, B., Cassai, E., and Nahmias, A. J. DNA Fragment of Herpes Simplex 2 and Its Transcription in Human Cervical Cancer Tissue. PNAS, 1972; 69: 3784-3789.
4: zur Hausen H, Schulte-Holthausen H, Klein G, Henle W, Henle G, Clifford P, Santesson L.EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature. 1970;228(5276):1056-8.
5: zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-6.
6: H Zur Hausen, H Schulte-Holthausen, H Wolf, K Dörries, H Egger. Attempts to detect virus-specific DNA in human tumors. II. Nucleic acid hybridizations with complementary RNA of human herpes group viruses. Int J Cancer. 1974, 13 (5): 657-664
7: zur Hausen H, Gissmann L, Steiner W, Dippold W, Dreger I. Human papilloma viruses and cancer. Bibl Haematol. 1975 ;(43):569-71.
8: zur Hausen H. Condylomata acuminata and human genital cancer. Cancer Res. 1976 Feb;36(2 pt 2):794.
9: Almeida, J. D., Oriel, J. D., and Stannard, L. M. Characterization of the Virus Found in Human Genital Warts. Microbios. 1969: 3: 225-232.
10: Dunn, A. E. G., and Ogilvie, M. M. Intranuclear Virus Particles in Human Genital Wart Tissue: Observations on the Ultrastructure of the Epidermal Layer J. Ultrastruct. Res.1968:22: 282-291.
11: Oriel, J. D., and Almeida, J. D., Demonstration of Virus Particles in Human Genital Warts. Brit. J. Venereal Disease. 1970:46: 37-42.
12: Siegel, A. Malignant Transformation of Condyloma Acuminatum. Am. J. Surg.,1962:103: 613-617.
13: Gissmann L, Hausen HZ. Human papilloma virus DNA: physical mapping and genetic heterogeneity. PNAS. 1976;73(4):1310-3.
14: Meisels A, Fortin R. Condylomatous lesions of the cervix and vagina. I. Cytologic patterns. Acta Cytol. 1976;20(6):505-9.
15: Meisels A, Roy M, Fortier M, Morin C, Casas-Cordero M, Shah KV, Turgeon H. Human papillomavirus infection of the cervix: the atypical condyloma. Acta Cytol. 1981;25(1):7-16.
16: Gissmann L, Pfister H, Zur Hausen H. Human papilloma viruses (HPV): characterization of four different isolates. Virology. 1977;76(2):569-80.
17: zur Hausen H. Human papillomaviruses and their possible role in squamous cell carcinomas. Curr Top Microbiol Immunol. 1977;78:1-30.
18: Gissmann L, zur Hausen H. Physical characterization of deoxyribonucleic acids of different human papilloma viruses (HPV). Med Microbiol Immunol. 1978;166(1-4):3-11.
19: Pfister H, zur Hausen H. Characterization of proteins of human papilloma viruses (HPV) and antibody response to HPV 1. Med Microbiol Immunol. 1978;166(1-4):13-9.
20: Pfister H, zur Hausen H. Seroepidemiological studies of human papilloma virus (HPV-1) infections. Int J Cancer. 1978;21(2):161-5.
21: Grussendorf EI, zur Hausen H. Localization of viral DNA-replication in sections of human warts by nucleic acid hybridization with complementary RNA of human papilloma virus Type 1. Arch Dermatol Res. 1979;264(1):55-63.
22: zur Hausen H. The role of viruses in human tumors. Adv Cancer Res. 1980;33:77-107.
23: Gissmann L, zur Hausen H. Partial characterization of viral DNA from human genital warts (Condylomata acuminata). Int J Cancer. 1980;25(5):605-9.
24: Pfister H, Nürnberger F, Gissmann L, zur Hausen H. Characterization of a human papillomavirus from epidermodysplasia verruciformis lesions of a patient from Upper-volta. Int J Cancer. 1981;27(5):645-50.
25: zur Hausen H, de Villiers EM, Gissmann L. Papillomavirus infections and human genital cancer. Gynecol Oncol. 1981;12:S124-8.
26: de Villiers EM, Gissmann L, zur Hausen H. Molecular cloning of viral DNA from human genital warts. J Virol. 1981;40(3):932-5.
27: Gissmann L, deVilliers EM, zur Hausen H. Analysis of human genital warts (condylomata acuminata) and other genital tumors for human papillomavirus type 6 DNA. Int J Cancer. 1982;29(2):143-6.
28: Gissmann L, Diehl V, Schultz-Coulon HJ, zur Hausen H. Molecular cloning and characterization of human papilloma virus DNA derived from a laryngeal papilloma. J Virol. 1982;44(1):393-400.
29: Schwarz E, Dürst M, Demankowski C, Lattermann O, Zech R, Wolfsperger E, Suhai S, zur Hausen H. DNA sequence and genome organization of genital human papillomavirus type 6b. EMBO J. 1983;2(12):2341- 8.
30: Gissmann L, Wolnik L, Ikenberg H, Koldovsky U, Schnürch HG, zur Hausen H. Human papillomavirus types 6 and 11 DNA sequences in genital and laryngeal papillomas and in some cervical cancers. PNAS. 1983 ;80(2):560-3.
31: Dürst M, Gissmann L, Ikenberg H, zur Hausen H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. PNAS. 1983;80(12):3812-5
32: Ikenberg H, Gissmann L, Gross G, Grussendorf-Conen EI, zur Hausen H. Human papillomavirus type-16- related DNA in genital Bowen’s disease and in Bowenoid papulosis. Int J Cancer. 1983;32(5):563-5.
33: Boshart M, Gissmann L, Ikenberg H, Kleinheinz A, Scheurlen W, zur Hausen H. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J. 1984;3(5):1151-7.
34: Tsunokawa Y, Takebe N, Nozawa S, Kasamatsu T, Gissmann L, zur Hausen H, TeradaM, Sugimura T. Presence of human papillomavirus type-16 and type-18 DNA sequences and their expression in cervical cancers and cell lines from Japanese patients. Int J Cancer. 1986;37(4):499-503.
35: Scheurlen W, Stremlau A, Gissmann L, Höhn D, Zenner HP, zur Hausen H. Rearranged HPV 16 molecules in an anal and in a laryngeal carcinoma. Int J Cancer. 1986;38(5):671-6.
36: zur Hausen H. Intracellular surveillance of persisting viral infections. Human genital cancer results from deficient cellular control of papillomavirus gene expression. Lancet. 1986 ;2(8505):489-91.
37: Boshart M, zur Hausen H. Human papillomaviruses in Buschke-Löwenstein tumors: physical state of the DNA and identification of a tandem duplication in the noncoding region of a human papillomavirus 6 subtype. J Virol. 1986 ;58(3):963-6.
38: Yee C, Krishnan-Hewlett I, Baker CC, Schlegel R, Howley PM. Presence and expression of human papillomavirus sequences in human cervical carcinoma cell lines. Am J Pathol. 1985;119(3):361-6.
39: Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370(9590):890-907.
40: Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues. Nat Rev Cancer. 2007 ;7(1):11-22.
41: zur Hausen H. Papillomaviruses in anogenital cancer as a model to understand the role of viruses in human cancers. Cancer Res. 1989;49(17):4677-81.
42: zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002 ;2(5):342-50.
43: Muñoz Net al. Variations in the age-specific curves of human papillomavirus prevalence in women worldwide. Int J Cancer. 2006;119(11):2677-84.
44: Strickler HD, Palefsky JM, Burk RD. HPV types present in invasive cervical cancers of HIV-seropositive women. Int J Cancer. 2008;123(5):1224-5.
45: Chin-Hong PV et al. Comparison of patient- and clinician-collected anal cytology samples to screen for human papillomavirus-associated anal intraepithelial neoplasia in men who have sex with men. Ann Intern Med. 2008;149(5):300-6.
46: Chang TC.et al. Cervical cancer screening program integrating Pap smear and HPV DNA testing: a population-based study. Int J Cancer. 2008;122(12):2835-41.
47: Schwarz E, Freese UK, Gissmann L, Mayer W, Roggenbuck B, Stremlau A, zur Hausen H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature. 1985;314(6006):111-4.
48: Mantovani F, Banks L. The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene. 2001;20(54):7874-87.
49: Duensing S, Münger K. Mechanisms of genomic instability in human cancer: insights from studies with human papillomavirus oncoproteins. Int J Cancer. 2004 ;109(2):157-62.
50: Wan F, Miao X, Quraishi I, Kennedy V, Creek KE, Pirisi L.Gene expression changes during HPV-mediated carcinogenesis: a comparison between an in vitro cell model and cervical cancer. Int J Cancer. 2008 ;123(1):32- 40.
51: Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990 ;248(4951):76-9.
52: Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63(6):1129-36.
53: Münger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumour suppressor gene product. EMBO J. 1989 ;8(13):4099-105.
54: Tong X, Howley PM. The bovine papillomavirus E6 oncoprotein interacts with paxillin and disrupts the actin cytoskeleton. PNAS, 1997;94(9):4412-7
55: Um SJ, Rhyu JW, Kim EJ, Jeon KC, Hwang ES, Park JS. Abrogation of IRF-1 response by high-risk HPV E7 protein in vivo. Cancer Lett. 2002;179(2):205-12.
56: Bachmann A, and zur Hausen H. Disturbance of tumor necrosis factor alpha-mediated beta interferon signaling in cervical carcinoma cells. J Virol. 2002;76(1):280-91.
57: von Knebel Doeberitz M, Bauknecht T, Bartsch D, zur Hausen H. Influence of chromosomal integration on glucocorticoid-regulated transcription of growth-stimulating papillomavirus genes E6 and E7 in cervical carcinoma cells. PNAS. 1991; 88(4):1411-5.
58: Dürst M, Bosch FX, Glitz D, Schneider A, zur Hausen H. Inverse relationship between human papillomavirus (HPV) type 16 early gene expression and cell differentiation in nude mouse epithelial cysts and tumors induced by HPV-positive human cell lines. J Virol. 1991;65(2):796-804.
59: Bosch FX, Schwarz E, Boukamp P, Fusenig NE, Bartsch D, zur Hausen H. Suppression in vivo of human papillomavirus type 18 E6-E7 gene expression in non tumorigenic HeLa X fibroblast hybrid cells. J Virol. 1990 ;64(10):4743-54.
60: Duensing S, Münger K. Mechanisms of genomic instability in human cancer: insights from studies with human papillomavirus oncoproteins. Int J Cancer. 2004;109(2):157-62.
61: Parkin, D. M. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006; 118(12): 3030-44.
62: Parkin DM, Bray F. Chapter 2: the burden of HPV-related cancers. Vaccine 2006;24: Suppl 3:S11-S25.
63: Breitburd F, Kirnbauer R, Hubbert NL, Nonnenmacher B, Trin-Dinh-Desmarquet C, Orth G, Schiller JT, Lowy DR. Immunization with viruslike particles from cottontail rabbit papillomavirus (CRPV) can protect against experimental CRPV infection. J Virol. 1995;69(6):3959-63.
64: Harro CD, Pang YY, Roden RB, Hildesheim A, Wang Z, Reynolds MJ, Mast TC, Robinson R, Murphy BR, Karron RA, Dillner J, Schiller JT, Lowy DR. Safety and immunogenicity trial in adult volunteers of a human papillomavirus 16 L1 virus-like particle vaccine. J Natl Cancer Inst. 2001 Feb 21;93(4):284-92
65: Garland SM, et al.Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. N Engl J Med. 2007;356(19):1928-43.
66: Paavonen J, et al.Efficacy of a prophylactic adjuvanted bivalent L1 virus-like-particle vaccine against infection with human papillomavirus types 16 and 18 in young women: an interim analysis of a phase III double-blind, randomised controlled trial. Lancet. 2007 ;369(9580):2161-70.
67: Roden R, Wu TC. How will HPV vaccines affect cervical cancer? Nat Rev Cancer. 2006;6(10):753-63. 68: Haug CJ. Human papillomavirus vaccination–reasons for caution. N Engl J Med. 2008;359(8):861-2
69: Gottlieb MS, Schroff R, Schanker HM and Saxon A. Pneumocystis pneumonia – Los Angeles. MMWR, Morb Mortal Wkly Rep. 1981; 30: 250-2.
70: CDC Task Force on Kaposi’s Sarcoma and Opportunistic Infections. Epidemiologic aspects of the current outbreak of Kaposi’s sarcoma and opportunistic infections. N Engl J Med. 1982; 306:248-52.
71: Gottlieb MS, Schroff R, Schanker HM, and Saxon A. Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexual men: evidence of a new acquired cellular immunodeficiency. N Engl J Med. 1981;305:1425-31.
72: Siegal FP, Lopez C, Hammer GS, et al. Severe acquired immunodeficiency in male homosexuals, manifested by chronic perianal ulcerative herpes simplex lesions. N Engl J Med. 1981;305: 1439-44.
73: Masur H, Michelis MA, Greene JB, et al. An outbreak of community acquired Pneumocystis carinii pneumonia : initial manifestation of cellular immune dysfunction. N Engl J Med. 1981; 305:1431-44.
74: Hymes KB, Cheung T, Greene JB, and Laubinstein L. Kaposi’s sarcoma in homosexual men: a report of eight cases. Lancet 1981; 2: 598-600.
75: Rozenbaum W. Multiple opportunistic infections in a male homosexual in France. Lancet. 1982; 6;1(8271):572-3.
76: Francis DP, Curran JW, Essex M. Epidemic acquired immune deficiency syndrome: epidemiologic evidence for a transmissible agent. J Natl Cancer Inst. 1983;71(1):1-4.
77: Baltimore D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature. 1970;226(5252):1209-11.
78: Temin HM, Mizutani S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature. 1970;226(5252):1211-3.
79: Rho HM, Poiesz B, Ruscetti FW, Gallo RC. Characterization of the reverse transcriptase from a new retrovirus (HTLV) produced by a human cutaneous T-cell lymphoma cell line. Virology. 1981;112(1):355-60.
80: Gallo RC, Popovic M et al. Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS). Science. 1983; 220(4599):865-7.
81: Barré-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vézinet-Brun F, Rouzioux C, Rozenbaum W, Montagnier L. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science. 1983; 220(4599):868-71.
82: Vilmer E, Barre-Sinoussi F, Rouzioux C, Gazengel C, Brun FV, Dauguet C, Fischer A, Manigne P, Chermann JC, Griscelli C, Montagnier L. Isolation of new lymphotropic retrovirus from two siblings with haemophilia B, one with AIDS. Lancet. 1984 ;1(8380):753-7.
83: Brun-Vezinet F, Rouzioux C, Barre-Sinoussi F, et al. Detection of IgG antibodies to lymphadenopathy-associated virus in patients with AIDS or lymphadenopathy syndrome. Lancet. 1984 ;1(8389):1253-6.
84: Kalyanaraman VS, Cabradilla CD, Getchell JP et al. Antibodies to the core protein of lymphadenopathy-associated virus (LAV) in patients with AIDS. Science. 1984;225(4659):321-3.
85: Brun-Vézinet F, Rouzioux C, Montagnier L, Chamaret S, Gruest J, Barré-SinoussiF, Geroldi D, Chermann JC, McCormick J, Mitchell S, et al. Prevalence of antibodies to lymphadenopathy-associated retrovirus in African patients with AIDS. Science. 1984;226(4673):453-6.
86: Vilmer E, Fischer A, Griscelli C, Barre-Sinoussi F, Vie V, Chermann JC, Montagnier L, Rouzioux C, Brun- Vezinet F, Rosenbaum W. Possible transmission of a human lymphotropic retrovirus (LAV) from mother to infant with AIDS. Lancet. 1984 ;2(8396):229-30
87: Papaevangelou G, Barre-Sinoussi F, Chermann JC, Montagnier L et al. Lymphadenopathy associated virus in AIDS, lymphadenopathy associated syndrome, and classic Kaposi patients in Greece. Lancet. 1984 ;2 (8403) :642-44.
88: Feorino PM, Francis D et al. Lymphadenopathy associated virus infection of a blood donor-recipient pair with acquired immunodeficiency syndrome. Science 1984 ; 225(4657): 69-72.
89: Klatzmann D, Barré-Sinoussi F, Montagnier L, et al. Selective tropism of lymphadenopathy associated virus (LAV) for helper-inducer T lymphocytes. Science. 1984;225(4657):59-63.
90: Montagnier L and F Barré-Sinoussi et al. Human T cell leukaemia lymphoma viruses (eds Gallo RC & Essex M) 1984; page 376-379. Cold Spring Harbour Laboratories, New York.
91: Chermann JC, Barré F, Montagnier L. Retroviruses and acquired immunodeficiency syndrome (AIDS). Bull Acad Natl Med. 1984 ;168(1-2):288-95.
92: Rey MA, Spire B, Dormont D, Barre-Sinoussi F, Montagnier L, Chermann JC. Characterization of the RNA dependent DNA polymerase of a new human T-lymphotropic retrovirus (lymphadenopathy associated virus). Biochem Biophys Res Commun. 1984;121(1):126-33.
93: Gallo RC, Salahuddin SZ, Popovic M, et al. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science. 1984: 224(4648): 500-3.
94: Popovic M, Sarngadharan MG, Read E, Gallo RC. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science. 1984: 224(4648): 497-500. 95: Schüpbach J, Popovic M, Gilden RV, Gonda MA, Sarngadharan MG, Gallo RC. Serological analysis of a subgroup of human T-lymphotropic retroviruses (HTLV-III) associated with AIDS. Science. 1984; 224(4648): 503-5.
96: Sarngadharan MG, Popovic M, Bruch L, Schüpbach J, Gallo RC. Antibodies reactive with human T- lymphotropic retroviruses (HTLV-III) in the serum of patients with AIDS. Science. 1984 ; 224(4648): 506-8.
97: Levy JA, Hoffman AD, Kramer SM, Landis JA, Shimabukuru JM, Oshiro LS. Isolation of lymphocytopathic retroviruses from San Francisco patients with AIDS. Science. 1984; 225(4664): 840-2.
98: Safai B, Sarngadharan MG, Groopman JE, Arnett K, Popovic M, Sliski A, Schüpbach J, Gallo RC. Seroepidemiological studies of human T-lymphotropic retrovirus type III in acquired immunodeficiency syndrome. Lancet. 1984;1(8392):1438-40.
99: Laurence J, Brun-Vezinet F, Barré-Sinoussi F, Chermann JC, Montagnier L et al. Lymphadenopathy- associated viral antibody in AIDS. Immune correlations and definition of a carrier state. N Engl J Med. 1984 ;311(20):1269-73.
100: Lee TH, , Haseltine W, Essex M et al. Serological cross-reactivity between envelope gene products of type I and type II human T-cell leukemia virus. PNAS. 1984 ;81(23):7579-83
101: Allan JS, Haseltine WA, Essex M et al. Major glycoprotein antigens that induce antibodies in AIDS patients are encoded by HTLV-III. Science. 1985;228(4703):1091-4.
102: Cheingsong-Popov R, Weiss RA, Dalgleish A, et al. Prevalence of antibody to human T-lymphotropic virus type III in AIDS and AIDS-risk patients in Britain. Lancet. 1984;2(8401):477-80
103: Kaminsky LS, Levy JA et al. High prevalence of antibodies to acquired immune deficiency syndrome (AIDS)-associated retrovirus (ARV) in AIDS and related conditions but not in other disease states. PNAS. 1985 ;82(16):5535-9.
104: Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, Gluckman JC, Montagnier L. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature. 1984 2;312(5996):767- 8.
105: Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984; 2;312(5996):763-7.
106: Hahn BH, Shaw GM, Arya SK, Popovic M, Gallo RC, Wong-Staal F. Molecular cloning and characterization of the HTLV-III virus associated with AIDS. Nature. 1984 ;312(5990):166-9.
107: Alizon M, Sonigo P, Barré-Sinoussi F, Chermann JC, Tiollais P, Montagnier L, Wain-Hobson S. Molecular cloning of lymphadenopathy-associated virus. Nature. 1984 ;312(5996):757-60.
108: Wain-Hobson S, Alizon M, Montagnier L. Relationship of AIDS to other retroviruses Nature. 1985 ;313(6005):743.
109: Coffin J, Haase A,Levy JA, Montagnier L, Temin HM and Varmus HJ, What to call the AIDS virus? Nature, 1986,321:10-12
110: Ratner L, Gallo RC, Wong-Staal F. HTLV-III, LAV, ARV are variants of same AIDS virus. Nature. 1985 ;313 (6004):636-7.
111: Keele BF, Van Heuverswyn F, Li Y, et al. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science 2006; 313:523-6.
112: Michael Worobey et al. Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature. 2008;455, 661-664.
113: Korber B, Muldoon M, Theiler J, et al. Timing the ancestor of the HIV-1 pandemic strains. Science. 2000;288:1789-96.
114: Van Heuverswyn F, Li Y, Neel C, et al. Human immunodeficiency viruses: SIV infection in wild gorillas. Nature 2006; 444:164.
115: Zhu T, Korber BT, Nahmias AJ, Hooper E, Sharp PM, Ho DD. An African HIV-1 sequence from 1959 and implications for the origin of the epidemic. Nature. 1998; 391:594-7.
116: Gilbert MT, Rambaut A, Wlasiuk G, Spira TJ, Pitchenik AE, Worobey M. The emergence of HIV/AIDS in the Americas and beyond. PNAS. 2007 ;104(47):18566-70.
117: Taylor BS, Sobieszczyk ME, McCutchan FE, Hammer SM. The challenge of HIV-1 subtype diversity. N Engl J Med. 2008;358(15):1590-602.
118: Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. Molecular architecture of native HIV-1 gp120 trimers. Nature. 2008;455(7209):109-13.
119: Hladik F, McElrath MJ. Setting the stage: host invasion by HIV. Nat Rev Immunol. 2008;8(6):447-57.
120: Haase AT. Perils at mucosal front lines for HIV and SIV and their hosts. Nat Rev Immunol. 2005 ;5(10):783-92.
121: Fraser, C., Hollingsworth, T.D., Chapman, R., de Wolf, F., Hanage, W.P. Variation in HIV-1 set-point viral load: epidemiological analysis and an evolutionary hypothesis. PNAS, 2007, 104:17441-17446.
122: Pilcher CD, et al. Amplified transmission of HIV-1: comparison of HIV-1 concentrations in semen and blood during acute and chronic infection. AIDS. 2007;21(13):1723-30.
123: McCune, J.M. 2001. The dynamics of CD4+ T-cell depletion in HIV disease. Nature. 410:974-979.
124: Brenchley, J.M., et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 2004: 200:749-759.
125: Hunt PW, et al. Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J Infect Dis. 2008;197(1):126- 33.
126: Miura T, Walker BD et al. Genetic characterization of human immunodeficiency virus type 1 in elite controllers: lack of gross genetic defects or common amino acid changes. J Virol. 2008;82(17):8422-30.
127: Ahuja SK. CCL3L1-CCR5 genotype improves the assessment of AIDS Risk in HIV-1-infected individuals. PLoS ONE. 2008;3(9):e3165.
128: Goulder PJ, Watkins DI. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol. 2008;8(8):619-30
129: Greene WC,et al. Novel targets for HIV therapy. Antiviral Res. 2008 Sep 11.
130: Bailey AC, Fisher M. Current use of antiretroviral treatment. Br Med Bull. 2008;87:175-92.
131: ammer SM, et al. International AIDS Society-USA. Antiretroviral treatment of adult HIV infection: 2008 recommendations of the International AIDS Society-USA panel. JAMA. 2008;300(5):555-70.
132: Moatti JP, Marlink R, Luchini S, Kazatchkine M. Universal access to HIV treatment in developing countries: going beyond the misinterpretations of the ‘cost-effectiveness’ algorithm. AIDS. 2008;22 Suppl 1:S59-66.
133: Chen TK, Aldrovandi GM. Review of HIV antiretroviral drug resistance. Pediatr Infect Dis J. 2008 ;27(8):749-52.
134: UNAIDS. 2007. AIDS epidemic update: December 2007 .http://www.unaids.org/en/KnowledgeCentre /HIVData/EpiUpdate/EpiUpdArchive/2007/default.asp
135: Antiretroviral therapy for HIV infection in adults and adolescents: recommendations for a public health approach. 2006, WHO. Geneva, Switzerland. http://www.who.int/hiv/pub/guidelines/adult/en/index.html
136: Towards universal access: scaling up priority HIV/AIDS interventions in the health sector. Progress report. 2007, WHO. Geneva, Switzerland. http://www.who.int/hiv/mediacentre/universal_access_progress_report_en.pdf .
137: Stover, J., et al. The global impact of scaling up HIV/AIDS prevention programs in low- and middle- income countries. Science. 2006;311:1474-1476
138: Takeuchi H, Matano T. Host factors involved in resistance to retroviral infection. Microbiol Immunol. 2008 ;52(6):318-25.
139: Ulenga NK, Sarr AD, Thakore-Meloni S, Sankalé JL, Eisen G, Kanki PJ. Relationship between human immunodeficiency type 1 infection and expression of human APOBEC3G and APOBEC3F. J Infect Dis. 2008 ;198(4):486-92.
140: Goila-Gaur R, Strebel K. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology. 2008;5:51-55.
141: Sheehy, A.M., Gaddis, N.C., Chol, J.D., Malim, M.H. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 418:646-650.
142: Stremlau, M., et al. 2004. The cytoplasmic body component TRIM5a restricts HIV-1 infection in Old World monkeys. Nature. 427:848-853.
143: Kumar A, Jeang KT.Insights into cellular microRNAs and human immunodeficiency virus type 1 (HIV-1). J Cell Physiol. 2008;216(2):327-31.
144: White RG, Glynn JR, Orroth KK, Freeman EE, Bakker R, Weiss HA, Kumaranayake L,Habbema JD, Buvé A, Hayes RJ. Male circumcision for HIV prevention in sub-Saharan Africa: who, what and when? AIDS. 2008 ;22(14):1841-50
145: Fauci AS, Greene WC et al. HIV vaccine research: the way forward. Science. 2008;321(5888):530-2
© The Nobel Committee for Physiology or Medicine 2008
Nobel Prizes and laureates
Six prizes were awarded for achievements that have conferred the greatest benefit to humankind. The 12 laureates' work and discoveries range from proteins' structures and machine learning to fighting for a world free of nuclear weapons.
See them all presented here.

