Advanced information
Scientific background:
Mature cells can be reprogrammed to become pluripotent (pdf)
Scientific background
Mature cells can be reprogrammed to become pluripotent
The 2012 Nobel Prize in Physiology or Medicine is awarded to Dr. John B. Gurdon and Dr. Shinya Yamanaka for their discovery that mature, differentiated cells can be reprogrammed to a pluripotent stem cell state. This represents a paradigm shift in our understanding of cellular differentiation and of the plasticity of the differentiated state. Cellular differentiation appears as a unidirectional process, where undifferentiated cells mature to various specialised cell fates, such as neurons, muscle and skin cells. The prevalent view during the first half of the 20th century was that the mature cells were permanently locked into the differentiated state, and unable to return to a fully immature, pluripotent stem cell state. In 1962, John B. Gurdon radically changed this view by demonstrating that the nucleus from a differentiated frog intestinal epithelial cell was capable of generating a fully functional tadpole upon transplantation to an enucleated egg. This discovery shattered the dogma that cellular differentiation could only be a unidirectional process. Gurdon’s discovery was the starting point for cloning endeavours in various organisms. However, the question remained whether an intact differentiated cell could be fully reprogrammed to become pluripotent. In 2006, by an astonishingly simple procedure, Shinya Yamanaka proved that introduction of a small set of transcription factors into a differentiated cell was sufficient to revert the cell to a pluripotent state. The resulting cells were called induced pluripotent stem (iPS) cells. Together, Gurdon and Yamanaka have transformed our understanding of cellular differentiation. They have demonstrated that the usually very stable differentiated state can be unlocked because it harbours a potential for reversion to pluripotency. This discovery has introduced fundamentally new research areas, and offers exciting new opportunities to study disease mechanisms.
Introduction
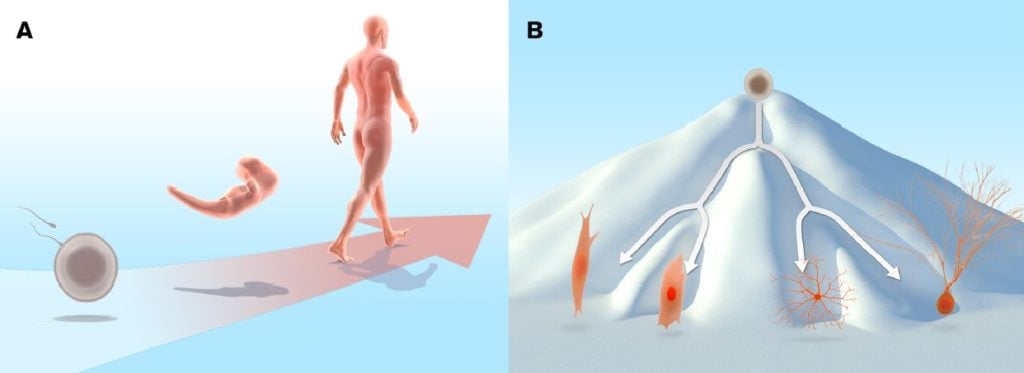
During normal development, cells proceed from the initial undifferentiated state of the egg and cells in the early embryo to a more specialised state. In the adult organism a range of differentiated cell types are required to execute the specialised functions performed in the adult body (Figure 1A). The fertilised egg and the cells in the early zygote are totipotent, in other words, they can give rise to all cell types in the embryo, as well as to extraembryonic tissues such as placenta. As development progresses, cells at the blastocyst stage start to become distinguishable: the inner cell mass gives rise to the embryo proper, whereas the surrounding cells make up the trophoblast lineage and are the source of extraembryonic tissues. The cells in the inner cell mass are pluripotent, i.e. they can give rise to all somatic cells, as well as to the germ cell lineage: the cells destined to become gametes (eggs and sperm).
During this developmental journey, cells progressively become more restricted in their differentiation potential and as a consequence, they do not retain pluripotency. Most cells mature into fully differentiated cells, although stem cells with limited potency remain in certain locations in the body and serve as a source for cell replacement, for example in the bone marrow, intestine and skin. Differentiated cells are remarkably stable and as a rule they will not shift fate into other types of differentiated cells or revert to the type of undifferentiated cells that can be found in the early embryo. For this reason, the long-standing predominant view was that cells in the somatic lineage were permanently in a locked state, such that the journey back to a highly undifferentiated state was impossible. The insight that various differentiated cell types were endowed with a specific pattern of proteins suggested that differentiated cells may carry irreversible epigenetic modifications or genetic alterations that render induction of pluripotency impossible. Conrad Hal Waddington proposed an epigenetic landscape of mountains and valleys as a metaphor for development. In this landscape, undifferentiated cells, represented as marbles, reside on a mountain top. During differentiation they trickle down into energetically more stable valleys, where they come to rest as differentiated cells. The assumption was that it would be difficult to revert the differentiated cells back to the undifferentiated state by moving them back to the mountain top (Waddington, 1957) (Figure 1B). Despite the dogma, the notion that specialised cells could somehow be unlocked from their differentiated state and dedifferentiate was not entirely dismissed. Various strategies to experimentally address the problem were considered. For example, Hans Spemann (Nobel Prize in Physiology or Medicine 1935) entertained the idea of transferring nuclei from differentiated cells to an immature cytoplasmic milieu to test its developmental potential. He referred to this approach as a “fantastic experiment”.
Reprogramming a differentiated somatic cell nucleus
A direct attempt to test whether differentiated cells in the somatic cell lineage were endowed with a dormant dedifferentiation potential was first carried out by Robert Briggs and Thomas King, who developed a technology for transfer of somatic cell nuclei from undifferentiated and differentiated cells to an enucleated fertilized egg in the amphibian Rana pipiens (Briggs and King, 1952). Amphibians are particularly amenable to this type of experiments because of the large size of the egg and the extrauterine development of embryos. Briggs and King showed that an embryonic nucleus, when transferred to an enucleated egg, could indeed support development up to the tadpole stage. In contrast, when they repeated the procedure with nuclei from more differentiated cells, they failed to obtain developing embryos. Thus, they concluded that differentiated nuclei undergo irreversible changes during differentiation such that the capacity to promote development was lost (King and Briggs, 1955).
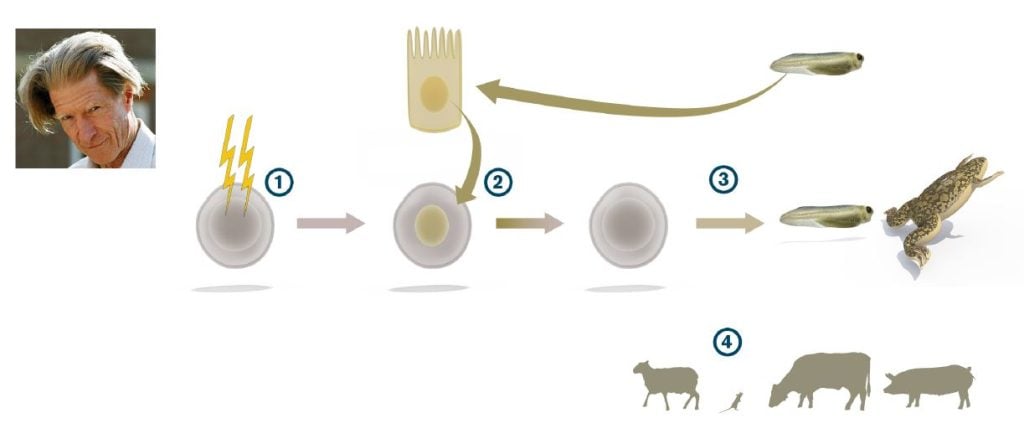
John B. Gurdon, who had trained in embryology with Michael Fischberg in Oxford, used a different amphibian, Xenopus laevis rather than Rana pipiens, to address the topic. In Xenopus, Gurdon could take advantage of a cell tracing system developed by Fischberg and colleagues (Elsdale et al., 1958) that allowed him to unequivocally distinguish the cells derived from transplanted nuclei from the cells of the host embryo. In a key study, Gurdon enucleated eggs by ultraviolet irradiation and found that when the eggs were transplanted with nuclei from differentiated tadpole intestinal epithelium, a small number of swimming tadpoles were indeed generated (Gurdon, 1962) (Figure 2). He could also show that the efficiency of nuclear reprogramming could be greatly improved by performing serial transplantations. By this strategy, he showed that a rather large proportion of all intestinal epithelial cell nuclei could be reprogrammed (Gurdon, 1962). Gurdon concluded that differentiated somatic cell nuclei had the potential to revert to pluripotency. However, considerable time passed before his discovery gained broad acceptance in the scientific community. In subsequent experiments, Gurdon used nuclei from adult frogs to generate tadpoles, and conversely, embryonic differentiated nuclei supported development of adult frogs (Gurdon and Uehlinger, 1966; Laskey and Gurdon, 1970).
Gurdon’s discovery was a fundamental paradigm shift, showing for the first time that the cell nucleus from a somatic differentiated cell was endowed with the capacity to drive development into a full range of somatic cell types and tissues after being placed in the cytoplasmic milieu of an egg cell.
Further developments of reprogramming by nuclear transfer
Gurdon’s discovery introduced a new research field centered on somatic cell nuclear transfer (SCNT) as a method to understand reprogramming and how cells change as they become specialized. In 1997, the first cloned mammal, the sheep Dolly, was born after SCNT from an adult mammary epithelial cell into an enucleated sheep egg (Wilmut et al., Nature 1997). The experimental strategy by Ian Wilmut and Keith Campbell was based on Gurdon’s work in Xenopus, but with additional technical adaptation. For example, one important modification was that nuclei used for transplantation in mammals came from mammary gland epithelial cells induced to enter quiescence, which make them better suited to synchronize with the early developing embryo. Since the cloning of sheep in 1997, SCNT has now been used to clone a plethora of mammalian species, including mouse, cow, pig, wolf and African wildcats. By nuclear transfer of cell nuclei from B- and T-cells in the immune system, conclusive evidence was provided that a differentiated cell nucleus with rearranged immunoglobulin or T-cell receptor genes could indeed be reprogrammed to support the development of a mouse (Hochedlinger and Jaenisch, 2002).
Reprogramming of an intact somatic differentiated cell to become pluripotent
Gurdon revealed that a differentiated cell nucleus has the capacity to successfully revert to an undifferentiated state, with a potential to restart development. However an open question remained, namely, whether it would be possible to induce reversion of an intact differentiated cell to a highly immature state. Many scientists considered this impossible, or at the very least, that it would require very complex reorganization in the cell to unlock the differentiated state. Such was the scene when Shinya Yamanaka decided to approach the problem of reprogramming to pluripotency. Yamanaka, who had trained both in orthopaedic surgery and molecular biology, became interested in the pluripotent state in part by studying pluripotent embryonic stem (ES) cells, first cultured and characterised by Martin Evans (Nobel Prize in Physiology or Medicine 2007).
Yamanaka’s laboratory focused on factors important for maintaining pluripotency in ES cells, such as ERas (Takahashi et al., 2003) and identified, in parallel with Austin Smith’s laboratory, the pluripotency gene Nanog (Mitsui et al., 2003; Chambers et al., 2003). Yamanaka then embarked on the quest of inducing pluripotency in somatic cells. From his work and others, he knew a large number of transcription factors that were expressed in ES cells with either confirmed or suspected functions in the maintenance of the pluripotent state. Furthermore, ES cells were known to induce pluripotency in somatic cell nuclei after induced cell fusions between ES and somatic cells (Tada et al., 2001). Equipped with this information, Yamanaka selected a set of 24 ES cell transcription factors that he considered as candidates to reinstate pluripotency in somatic cells.
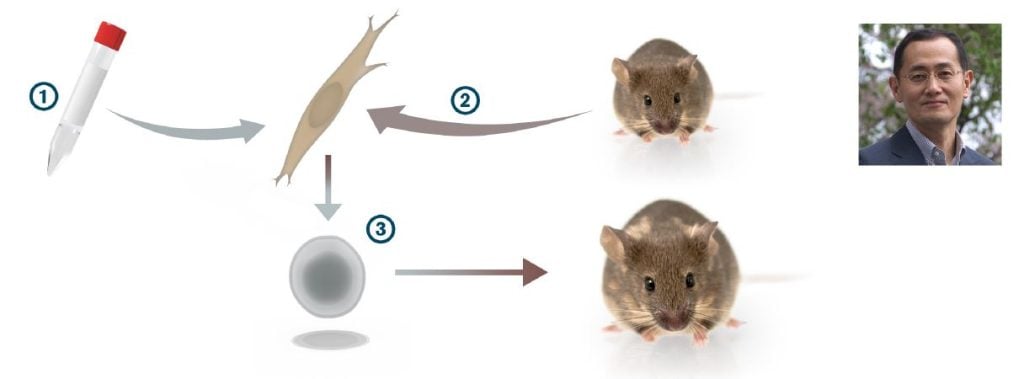
In a strikingly bold experiment, all 24 genes encoding these transcription factors were introduced in one step into skin fibroblasts and a few of them actually generated colonies that showed a remarkable resemblance to ES cells. The number of genes capable of inducing such colonies were reduced, one-by-one, to identify a combination of only four transcription factors (Myc, Oct3/4, Sox2 and Klf4) that were sufficient to convert mouse embryonic fibroblasts to pluripotent stem cells (Takahashi and Yamanaka, 2006) (Figure 3). The pluripotent stem cells, which Yamanaka called induced pluripotent stem cells (iPS cells), appeared with a very low frequency, but could be selected for by expression of a neomycin/lacZ fusion gene (bgeo) inserted into the Fbx15 locus in the genome of the mouse from which the fibroblasts were obtained. The Fbx15 promoter is active in pluripotent stem cells, and activation of bgeo expression in the pluripotent stem cells results in G418 resistance. The iPS cells generated various cell types in teratoma assays and contributed to tissues in chimeric mice after injection in mouse blastocysts. However, iPS cell germ line transmission was not achieved in the first study (Takahashi and Yamanaka, 2006). However, a year later, Yamanaka’s group, in parallel with Rudolph Jaenisch’s and Konrad Hochedlinger’s groups, refined the selection system (now selecting for activation of either the Oct4 or Nanog gene locus), and the resulting iPS cells showed germ line transmission (Okita et al., 2007; Wernig et al., 2007; Maherali et al., 2007). In 2007, Yamanaka’s and James Thomson’s laboratories were the first to produce human iPS cells (Takahashi et al., 2007; Yu et al., 2007). In the human iPS cell experiments, Yamanaka used the four factor combination from the 2006 paper (Myc, Oct4, Sox2 and Klf4), whereas Thomson used a somewhat different transcription factor combination (Lin28, Nanog, Oct 4 and Sox2).
Yamanaka’s discovery of iPS cells represents a truly fundamental discovery, as it was the first time an intact differentiated somatic cell could be reprogrammed to become pluripotent. Yamanaka’s discovery has opened up a completely new research field, and the astonishingly simple iPS technology is now used in a large number of laboratories around the world.
Further developments of cellular reprogramming and its use in medical research
Since the initial discovery, the technology has been improved in several ways. For example, the pluripotency factors can now be delivered into the cell without the use of retroviral vectors, which integrate randomly in the genome and cause deregulation of nearby endogenous genes that may contribute to tumour formation. Non-integrating viruses, stabilised RNAs or proteins, as well as episomal plasmids, are now used for integration-free delivery of the pluripotency genes. In certain cell types fewer than four factors are required to induce pluripotency, for example adult mouse neural stem cells only require Oct4 for iPS cell induction (Kim et al., 2009). Similarly, small molecules have been shown, in certain cellular contexts, to substitute for some of the pluripotency factors (Li et al., 2009). Importantly, iPS cells meet the most stringent criteria for pluripotency, as they are able to generate all iPS cell-mice in tetraploid complementation tests, i.e., when the cells are introduced into a tetraploid 8-cell stage morula (Zhao et al., 2009). All these improvements, based on the original discovery by Yamanaka, have been important steps to make the iPS technology more efficient and useful.
Yamanaka’s discovery, demonstrating that dramatic changes in the usually very stable differentiated state can be achieved, has also inspired research efforts to change the fate of cells without proceeding through a pluripotent state. Transdifferentiation experiments have a long history that goes back to imaginal disc experiments in Drosophila in the 1960s, followed by the use of single genes such as Antennapedia, MyoD, GATA1 or Pax5 to induce cell fate switches. In particular the discovery that MyoD could transdifferentiate 10T1/2 fibroblasts to myoblasts (Davis et al., 1987) showed that genes that carried out transdifferentiation could be systematically identified. Yamanaka’s approach to systematically define a small set of transcription factors, rather than a single factor, has inspired a recent wave of transdifferentiation experiments using combinations of transcription factors. For example, exocrine cells convert to endocrine cells in the pancreas by introduction of three transcription factors (Zhou et al., 2008). Similarly, cardiomyocytes can be generated from fibroblasts in vitro by introducing three different transcription factors (Ieda et al., 2010), and a corresponding cell fate switch has recently been accomplished in vivo by using the same factors (Song et al., 2012; Qian et al., 2012). These examples provide evidence for transdifferentiation within a germ layer, but efficient transdifferentiation between germ layers can also be achieved. The transdifferentiation from fibroblasts (mesoderm) to neurons (ectoderm) (so called iN cells) was accomplished by expression of three transcription factors (Pang et al., 2011).
Stem cells, including iPS cells, can potentially be used to replace diseased or lost cells in degenerative disorders including Parkinson’s disease and in type 1 diabetes. Cell replacement therapy with iPS cells can allow autologous cell grafting that would be less prone to immune rejection. The improved methods for the generation of iPS cells will be important in these efforts. However, the possibility exists that currently used procedures for reprogramming may introduce mutations or other genomic abnormalities, which may render them unsuitable for cell therapy. The prospect of using pluripotent stem cells, including ES cells, in cell transplantation remains challenging for a number of additional reasons. Thus, although this is a very exciting and promising research area, further work is required to ensure that using cells originating from pluripotent stem cells in therapy is safe for patients.
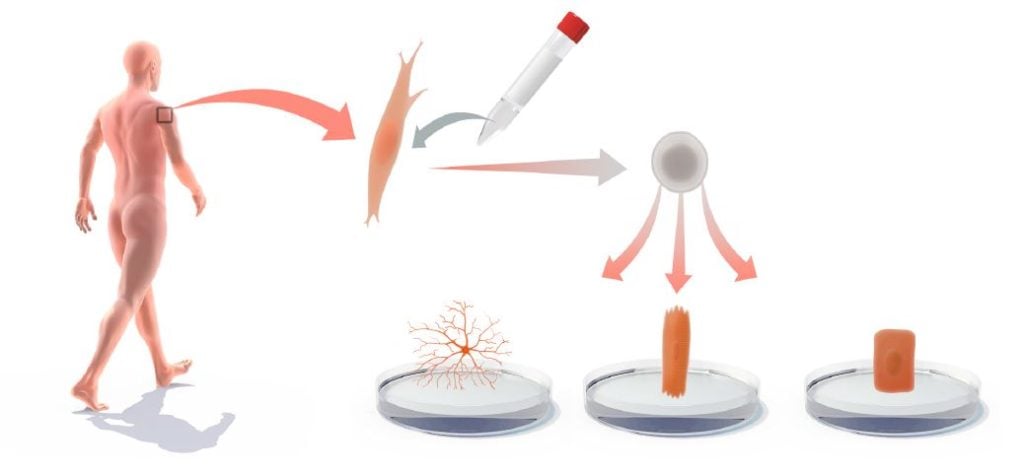
Another, more imminent, area for medical use is to derive iPS cells from patients with genetic and other disorders and then use the iPS cells for in vitro differentiation to gain novel insights into the disease process or to produce cell-based platforms for toxicology testing or drug development (Onder and Daley, 2012) (Figure 4). iPS cells have been produced from a large spectrum of diseases, including amyotrophic lateral sclerosis (ALS), Rett syndrome, spinal muscular atrophy (SMA), a1-antitrypsin deficiency, familial hypercholesterolemia and glycogen storage disease type 1A. For cardiovascular disease, there are now iPS cell models for Timothy syndrome, LEOPARD syndrome, as well as type 1 and 2 long QT syndrome (Onder and Daley, 2012). In several of these iPS cell-based disease models, disease‐relevant phenotypes are observed. For example, a progressive loss of motor neurons is observed in the iPS model for SMA. Moreover, Rett syndrome-specific iPS cells show reduced spine density after neuronal differentiation (Marchetto et al., 2010). Hepatocytic differentiation of iPS cells from a1-antitrypsin-deficient patients leads to elevated lipid and glycogen accumulation (Rashid et al., 2010). In vitro-differentiated iPS cell models can also mimic aspects of diseases with late onset, such as Alzheimer’s disease (Israel et al., 2012), Spinocerebellar ataxia (Koch et al., 2011) and Huntington’s disease (The HD iPSC Consortium, 2012). Progress has also been made when it comes to modelling diseases with complex genetics, such as schizophrenia (Brennand et al., 2011). However there are also diseases, for which it may be difficult to successfully mimic pathology in cultured iPS cell‐derived cells. Furthermore, for some diseases, notably in the hematopoietic lineage, robust in vitro differentiation protocols for iPS cells are lacking, limiting progress in this area.
iPS cell-based in vitro differentiated cells are also increasingly used as screening platforms for development and validation of therapeutic compounds. In an iPS cell-based model for familial dysautonomia, a novel compound, kinetin, was identified, which could partially reverse the aberrant splicing of the IKBKAP gene that causes the disease (Lee et al., 2009). Similarly, in a Long QT syndrome iPS cell model, beta blockers and ion channel blockers proved effective in modulating the phenotype (Itzhaki et al., 2011). iPS cells are thus becoming valuable new tools to gain insight into disease processes and they are now used to test and validate new therapeutics. Furthermore, even diseases with complex genetics and late onset can be successfully modelled by this “disease in the dish” approach.
In summary, the concept that mature, differentiated cells can be reprogrammed to a pluripotent stem cell state is a paradigm-shifting discovery. This insight has influenced essentially all areas of medicine or physiology. The discoveries made by John Gurdon and Shinya Yamanaka clearly stand out as truly fundamental and have introduced an entirely new research field.
Jonas Frisén, Urban Lendahl and Thomas Perlmann
References
Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S et al (2011). Modelling schizophrenia using human induced pluripotent stem cells. Nature 473: 221-225.
Briggs R, King TJ (1952). Transplantation of Living Nuclei from Blastula Cells into Enucleated Frog’s Eggs. Proc Natl Acad Sci USA 38: 455-463.
Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S et al (2003). Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 113: 643-655.
Davis RL, Weintraub H, Lassar AB (1987). Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51: 987-1000.
Elsdale TR, Fischberg M, Smith S (1958). A Mutation That Reduces Nucleolar Number in Xenopus-Laevis. Exp Cell Res 14: 642-643.
Gurdon JB (1962). Developmental Capacity of Nuclei Taken from Intestinal Epithelium Cells of Feeding Tadpoles. J Embryol Exp Morph 10: 622‐640.
Gurdon JB, Uehlinger V (1966). “Fertile” intestine nuclei. Nature 210: 1240‐1241.
Hochedlinger K, Jaenisch R (2002). Monoclonal mice generated by nuclear transfer from mature B and T donor cells. Nature 415: 1035‐1038.
Ieda M, Fu JD, Delgado‐Olguin P, Vedantham V, Hayashi Y, Bruneau BG et al (2010). Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142: 375‐386.
Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C et al (2012). Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482: 216‐220.
Itzhaki I, Maizels L, Huber I, Zwi‐Dantsis L, Caspi O, Winterstern A et al (2011). Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471: 225-229.
Kim JB, Greber B, Arauzo-Bravo MJ, Meyer J, Park KI, Zaehres H et al (2009). Direct reprogramming of human neural stem cells by OCT4. Nature 461: 649‐643.
King TJ, Briggs R (1955). Changes in the Nuclei of Differentiating Gastrula Cells, as Demonstrated by Nuclear Transplantation. Proc Natl Acad Sci USA 41: 321‐325.
Koch P, Breuer P, Peitz M, Jungverdorben J, Kesavan J, Poppe D et al (2011). Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 480: 543‐546.
Laskey RA, Gurdon JB (1970). Genetic content of adult somatic cells tested by nuclear transplantation from cultured cells. Nature 228: 1332‐1334.
Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA et al (2009). Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 461: 402‐406.
Li W, Zhou H, Abujarour R, Zhu S, Young Joo J, Lin T et al (2009). Generation of human‐induced pluripotent stem cells in the absence of exogenous Sox2. Stem Cells 27: 2992‐3000.
Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K et al (2007). Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 1: 55‐70.
Marchetto MCN, Carromeu C, Acab A, Yu D, Yeo GW, Mu YL et al (2010). A Model for Neural Development and Treatment of Rett Syndrome Using Human Induced Pluripotent Stem Cells. Cell 143: 527‐539.
Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K et al (2003). The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 113: 631‐642.
Okita K, Ichisaka T, Yamanaka S (2007). Generation of germline‐competent induced pluripotent stem cells. Nature 448: 313-317.
Onder TT, Daley GQ (2012) New lessons learned from disease modeling with induced pluripotent stem cells. Curr Opin Genet Dev (epub ahead of print).
Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ et al (2011). Induction of human neuronal cells by defined transcription factors. Nature 476: 220‐223.
Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L et al (2012). In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485: 593‐598.
Rashid ST, Corbineau S, Hannan N, Marciniak SJ, Miranda E, Alexander G et al (2010). Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest 120: 3127‐3136.
Song KH, Nam YJ, Luo X, Qi XX, Tan W, Huang GN et al (2012). Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485: 599‐604.
Tada M, Takahama Y, Abe K, Nakatsuji N, Tada T (2001). Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr Biol 11: 1553‐1558.
Takahashi K, Mitsui K, Yamanaka S (2003). Role of ERas in promoting tumour‐like properties in mouse embryonic stem cells. Nature 423: 541‐545.
Takahashi K, Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: 663-676.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K et al (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861‐872.
The HD iPSC Consortium (2012) Induced pluripotent stem cells from patients with Huntington’s disease show CAG‐repeat‐expansion‐associated phenotypes. Cell Stem Cell 11: 264‐278.
Waddington CH (1957) The Strategy of the Genes; a Discussion of Some Aspects of Theoretical Biology. Allen & Unwin.
Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K et al (2007). In vitro reprogramming of fibroblasts into a pluripotent ES‐cell‐like state. Nature 448: 318‐324.
Wilmut I, Schnieke AE, McWhir J, Kind AJ, Campbell KHS (1997). Viable offspring derived from fetal and adult mammalian cells. Nature 385: 810‐813.
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S et al (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318: 1917‐1920.
Zhao XY, Li W, Lv Z, Liu L, Tong M, Hai T et al (2009). iPS cells produce viable mice through tetraploid complementation. Nature 461: 86-90.
Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA (2008). In vivo reprogramming of adult pancreatic exocrine cells to beta‐cells. Nature 455: 627‐630.
Nobel Prizes and laureates
Six prizes were awarded for achievements that have conferred the greatest benefit to humankind. The 12 laureates' work and discoveries range from proteins' structures and machine learning to fighting for a world free of nuclear weapons.
See them all presented here.

