Advanced information
Scientific background:
Discoveries concerning the genomes of extinct hominins and human evolution (pdf)
Scientific background
Discoveries concerning the genomes of extinct hominins and human evolution
The 2022 Nobel Prize in Physiology or Medicine is awarded to Svante Pääbo for his discoveries concerning the genomes of extinct hominins and human evolution. The relationship between Homo sapiens and extinct hominins has long been a topic of great interest. Paleontology and archeology are important for studies of human evolution. Modern DNA technology provides opportunities to investigate our ancient past with more precision. However, due to extreme technical challenges resulting from degradation of DNA during tens of thousands of years and contamination from micro-organisms and contemporary humans, it was long questionable whether the analysis of archaic DNA from extinct hominin forms would be possible. Through extensive technological developments, Svante Pääbo set new rigorous standards in this challenging area and succeeded in obtaining the genome sequence of our closest extinct relative, the Neanderthal. This was followed by his sensational discovery of another extinct hominin, the Denisova, entirely from genome data retrieved from a small finger bone specimen. Svante Pääbo’s work further established that Homo sapiens had mixed with Neanderthals and Denisovans during periods of co-existence, resulting in introgression of archaic DNA in present-day humans. Striking examples of archaic gene variants that influence the physiology of present-day humans have already been demonstrated in a research field that is now highly dynamic. Through his groundbreaking discoveries, Pääbo opened a new window to our evolutionary past, revealing an unexpected complexity in the evolution and ad-mixture of ancient hominins, as well as providing the basis for an improved understanding of genetic features that make us uniquely human.
The question of our origin has long intrigued humanity and remains the focus of intense interest and discussion. The evolution of all currently living organisms started billions of years ago with the first primates appearing 55-65 million years ago. It is estimated that the last common ancestor of Old World monkeys and great apes lived around 25 million years ago and that of humans and chimpanzees around 6 million years ago. The genus Homo evolved around 3 million years ago, with Homo erectus entering the scene 1.8 million years ago. Like most evolutionary processes, our early history is characterized by numerous genetic branches, many of which were ultimately unsuccessful. However, Homo erectus survived longer than any other hominin species and was the first known hominin to migrate out of Africa, where subsequent evolution led to the Homo neandertalensis, our archaic relatives, the Neanderthals.
For a long time, studies of human evolution relied on analyses of ancient bone remains, their morphological characteristics and the examination of tools and other archeological artifacts. The first skeletal remains identified as Neanderthal, were found in 1856 in the Neandertal valley in Germany. Since then, specimens from multiple Neanderthal individuals have been identified in locations spanning a vast geographical range in Eurasia. Neanderthals inhabited Europe and Asia from around 400,000 to 30,000 years ago, at which point they went extinct. Anatomically modern humans, Homo sapiens, with skeletons like those of present-day humans, first appeared in Africa around 300,000 years ago. Around 60,000-70,000 years ago, migration from Africa into the Middle East led to an expansion of Homo sapiens across Eurasia. Thus, Homo sapiens co-existed with Neanderthals in Eurasia for at least 20,000 years, probably longer. However, the nature of their interactions was a long-standing matter of debate. It became increasingly clear that genetic analysis was required to shed light on the relationship between us and our Neanderthal relatives.
The first steps to use genetic tools to study how contemporary humans are related to each other and to elucidate our evolutionary origin were taken by Allan Wilson. In a key study, he performed restriction enzyme analyses of mitochondrial DNA (mtDNA), the portion of human DNA that is most accessible since it is present at high copy numbers in each cell, from current African and non-African population groups [1]. His results indicated a common origin in Africa for all sub-populations of Homo sapiens. These results were foundational for the Out of Africa theory of human evolution. However, they were not definitive since mtDNA is only inherited on the maternal side and it comprises a mere 16,500 base pairs (bp) compared to 3 billion bp for the nuclear genome. Therefore, more comprehensive data, including from the nuclear human genome were required.
The springboard for studies of the human nuclear genome was the Human Genome Project, which was launched in 1990 by a large international consortium. The objective was to sequence and annotate a full haploid nuclear genome and to generate a human reference map for future studies. In 2001, two landmark papers were published describing the sequence of the human nuclear genome [2, 3], and remaining complex regions were completed as late as this year [4]. The reference genome is available to the public from multiple sources including University of California Santa Cruz, National Center for Biotechnology Information, GenBank and Ensembl. Additional human genomes from diverse population groups were subsequently reported, including those from the 1000 Genomes Project Consortium [5] and the Simons Genomes Diversity Project [6], offering critical resources for understanding human population genetics and exploring our evolutionary past. However, the question of how we are related to extinct hominins, such as Neanderthals, would require more than inference studies from contemporary human DNA. It would require the extraction and sequencing of very old DNA from an extinct species, a monumental task that seemed unachievable.
From a young age, and while studying medicine at Uppsala University, Svante Pääbo nurtured a strong interest in Egyptology. During graduate studies on adenoviruses and their interaction with the immune system in Per A. Peterson’s group, he secretly worked on a side project aiming to isolate DNA from mummy specimens. He managed to clone a DNA library from a 2,400-year-old mummy sample and screened it with human repeat sequences, revealing human DNA among the clones. However, Pääbo soon realized that working with ancient DNA is plagued with techno-logical challenges and he later acknowledged that the results described in the first publication likely suffered from contamination by DNA from contemporary humans. He therefore focused on improving the techniques, which was best done by analyzing ancient DNA from non-human species where contamination was more easily detected. In 1987, after a short fellowship in the laboratory of Walter Schaffner at the University of Zürich, Pääbo moved to the University of California, Berkeley to work on ancient DNA as a post-doctoral fellow in Allan Wilson’s lab.
Methodological challenges in the analysis of ancient DNA
In the Wilson lab, and during the next decades of his career, Svante Pääbo worked tirelessly to develop protocols for retrieving, sequencing, and analyzing DNA from archaic specimens. Although much of this work was performed on other species, his ultimate objective was to obtain sequence information from DNA extracted from Neanderthal bone. In the mid-1980s, the polymerase chain reaction (PCR), which was invented by Kary Mullis and awarded the Nobel Prize in Chemistry in 1993 [7], revolutionized the field and greatly facilitated the analysis of trace amounts of DNA. However, the risk of recovering contaminating DNA remained and even in-creased. In 1988, Pääbo and Wilson were the first to apply PCR to ancient remains [8]. Pääbo was acutely aware of the technical challenges associated with DNA analysis of very old bone samples. He and others had demonstrated that bones from the period of interest, the late Pleistocene, are heavily contaminated by DNA from microbes and contemporary humans handling the specimens, while endogenous DNA, if present at all, is found at trace amounts. Several groups in the field were active, but many studies suffered from contamination leading to artifactual results. Svante Pääbo decisively addressed this issue by designing specialized clean rooms, minimizing contamination, and emphasizing the need for independent reproduction of results by other laboratories.
Methods to determine how much ancient DNA was present in given specimens were also desperately needed, and a better understanding of the biochemical properties of ancient DNA was required. The type of DNA damage observed in ancient samples include fragmentation and modifications of nucleotides caused by oxidative processes such as deamination of cytosine residues to uracil. Over several decades, Pääbo’s group described the challenges associated with analyses of ancient DNA and provided solutions to these [9-11]. The group also developed methods for extracting DNA from ancient samples using silica-based purification methods [12] and demonstrated that the extent of racemization of certain amino acids can be used to determine if specimens contained endogenous DNA [13].
Studies of the theoretical decay rate were also important to understand the fate of old DNA. An article by Thomas Lindahl, whom Pääbo worked with for a short time at the Imperial Cancer Research Fund in London, argued that specimens up to tens of thousands and perhaps even hundred thousand years may be successfully analyzed, while older samples were expected to be unproductive [14]. However, the conditions under which the DNA is stored can greatly influence the decay rate with low temperature being less damaging. For example, the recent sequencing of DNA from a tooth of a million-year-old mammoth recovered from permafrost illustrates this point [15].
The discoveries
The initial breakthrough: Neanderthal mito-chondrial DNA
Despite the invention of PCR, it remained highly questionable whether the minute amounts of remaining DNA found in archaic bone could have resisted chemical modifications and fragmentation for tens of thousands of years, enough to withstand the competing more recent DNA from microbial growth and present-day human DNA contamination. Nevertheless, Svante Pääbo, who in 1990 was recruited to the University of Munich, was determined to reach this goal. The original finding of Neanderthal skeletal remains was made in Feldhofer cave, Neandertal valley near Düsseldorf in 1856. The remains of this “type specimen” is kept at the Rheinisches Museum in Bonn, Germany. When Pääbo set out on his ambitious quest to study the Neanderthal genome, this was where he first turned for access to the precious remains that he needed as a source of material. Thankfully, he was well received, and he obtained a piece of a Neanderthal humeral bone, on a collaborative basis.
The mtDNA has a greater chance of persisting in old skeletons than nuclear DNA since every cell contains hundreds to thousands of copies of mtDNA, but only two copies (one maternal and one paternal) of each segment of nuclear DNA. Therefore, Pääbo selected mtDNA as the first Neanderthal sequence target for his investigations. Two primers defining a 105 bp segment of the hypervariable part of the human mtDNA control region were used for PCR amplification and the fragments resulting from two independent experiments were cloned and sequenced. The initial mtDNA sequence he obtained was 61 nucleotides long. Most of the clones contained variants that had not been seen among present-day humans. Extensive precautions were taken, and control experiments were performed to ensure that the sequence was endogenous. The experiments were repeated from a new piece of bone with the same result. A piece of the Neanderthal bone was also sent to Mark Stoneking at the Anthropological Genetics laboratory, Pennsylvania State University, for replication in an independent laboratory. Over-lapping DNA fragments were then amplified, cloned, and sequenced to reconstruct a longer sequence. A total of 123 clones representing 13 fragments were sequenced at least twice each, expanding the Neanderthal mtDNA sequence to 379 nucleotides [16].
To investigate the relationship between modern day humans and Neanderthals, the 379 nucleotides of Neanderthal mtDNA were compared with a collection of 2051 human and 59 chimpanzee mtDNA sequences. The sequence was shown to fall outside the variation of European, African, Asian, Native American, and Australian/Oceanic Homo sapiens (Figure 1).
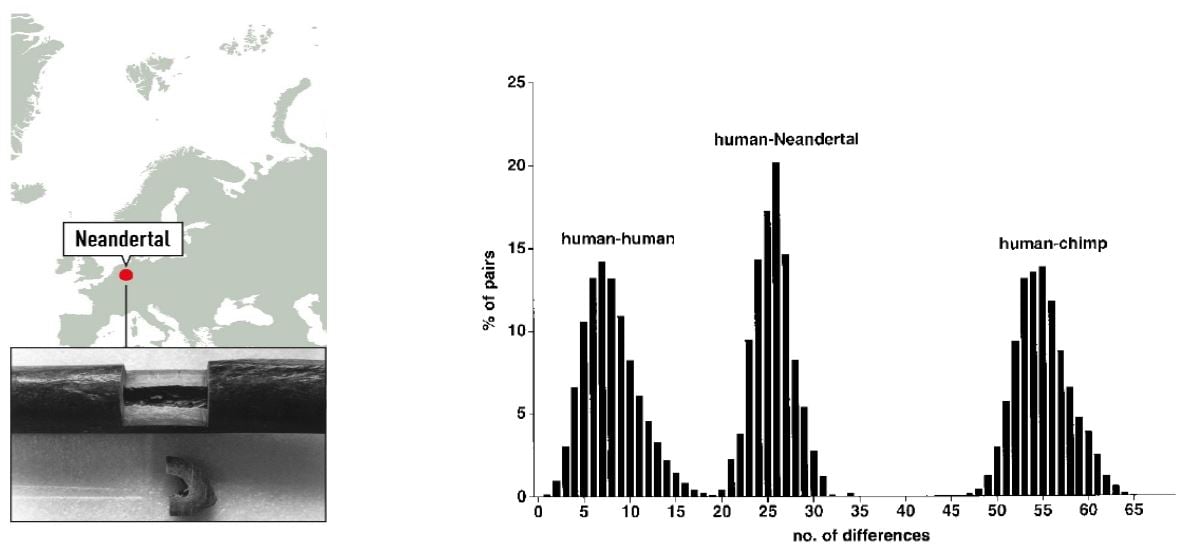
Figure 1. Left: Map of the Neandertal site and sample from the right humerus of the type specimen. Right: Distributions of Pairwise Sequence Differences among Humans, the Neandertal, and Chimpanzees (Krings et al. 1997).
Phylogenetic tree modelling indicated that the present-day mtDNA gene pool originates in Africa, consistent with Wilson’s previous findings, and that the common ancestor carrying this mtDNA lived 120,000 – 150,000 years ago. Based on the mtDNA analysis, the time since the existence of a common ancestor of Neanderthal and anatomically modern human was estimated to be four times greater; 550,000 to 690,000 years. The results suggested that Neanderthals went extinct without contributing mtDNA to present-day humans.
As mtDNA is exclusively inherited through the maternal line and only can be transmitted to the next generation by females, it can only give a limited picture of our evolutionary history. As it reflects the female side, mtDNA would only reveal if women moved between the two groups. Because of its strict maternal inheritance and lack of recombination, mtDNA variants can also disappear by chance, if carrier women only have sons. Thus, analyses of mtDNA are less informative than investigations of the nuclear genome. The results from sequencing and analysis of Neanderthal mtDNA did not exclude the possibility that Neanderthals contributed other genes to present-day humans. The sequencing of Neanderthal mtDNA proved that it is possible to sequence DNA from a 40,000-year-old bone from our most recent extinct relative. However, more bones were needed to consolidate and expand these exciting results.
Vindija in Croatia is known for significant findings of Neanderthal bones. Like the original Neandertal site in Germany, it contains limestone caves that are particularly suitable for preserving DNA due to their slightly basic chemical environment. The specimens are kept at the Institute of Quartenary Paleontology and Geology of the Croatian Academy of Science and Arts in Zagreb. Pääbo visited the Institute and was given small samples from 15 Neanderthal bones. Amino acid analysis indicated that seven specimens potentially contained Neanderthal DNA in sufficient quantities for analysis. The most promising sample was carbon-14 dated and shown to be 42,000 years old.
In 2000, a British group published the second Neanderthal mtDNA sequence, from the Mezmaiskaya cave in Northern Caucasus [17]. Pääbo’s group published the third mtDNA sequence from the Vindija specimen shortly thereafter, allowing estimations of genetic diversity among Neanderthals [18]. Allan Wilson had shown that chimpanzees, gorillas, and orangutans have a greater degree of genetic variation than modern humans even though they are much less abundant, indicating that Homo sapiens expanded from a smaller population. Analysis of the three Neanderthal mtDNA sequences gave a similar picture, indicating that these individuals came from a small population that had subsequently undergone expansion.
In 2004 Pääbo extended the study of mtDNA variation in Neanderthals to include an additional four Neanderthal samples originating from geographically disparate sites across Europe as well as five early Homo sapiens [19]. Based on this analysis of the mtDNA, the group concluded that it was highly unlikely that there was a large genetic contribution of Neanderthals to early modern humans, although they could not exclude the possibility of a smaller genetic contribution.
The first million Neanderthal nucleotides
Pääbo, who had now established the Max Planck Institute for Evolutionary Anthropology in Leipzig, Germany, turned to the nuclear genome, spurred by emergence of novel sequencing technology. Using a technique that was new at the time, offered by 454 Life Sciences, single-stranded DNA libraries flanked by common adapters, were created and individual molecules were amplified through bead-based emulsion PCR followed by pyrosequencing [20]. This sequencing platform provided several advantages over the PCR, cloning and sequencing approach previously used. Importantly, it generated hundreds of thousands of reads per run, thereby providing a high-throughput format. It also enabled direct sequencing of all DNA in a sample, without selecting sequences by PCR, and circumvented the need for bacterial cloning, in which large amounts of template molecules are lost. It further precluded template competition in PCR as each molecule was amplified in isolation. Its read lengths of 100-200 nucleotides were suitable for the average length of the DNA preserved in archaic bones. It also allowed deduction of the frequency of different nucleotide misincorporations, as each sequenced product stemmed from just one original single-stranded template molecule of known orientation.
Pääbo’s team tested more than 70 Neanderthal bone and tooth samples from different sites in Europe and western Asia to estimate bio-molecular preservation. Most samples were unlikely to contain retrievable endogenous DNA, but some contained high levels of amino acids and low levels of amino acid racemization, factors indicating that chances of retrieving archaic DNA were reasonable. The level of contamination with modern human DNA was assessed by PCR and sequencing of mtDNA. It was shown to be highly variable between the samples. One bone from Vindija cave, Croatia, stood out in that >90% of the mtDNA segments were of Neanderthal origin.
This bone was chosen for high-throughput sequencing. A total of 254,933 unique sequences were recovered and aligned to the human, chimpanzee, and mouse genome sequences and to GenBank databases using the program BLASTN. Sequences were analyzed whose closest matches were to the human or chimpanzee nuclear genomes of at least 30 base pairs long. Overall 0.04% of the Neanderthal nuclear genome sequence was covered by the reads.
The first million nucleotides generated by high-throughput sequencing were published in 2006 [21]. An alternative strategy based on cloning from the same bone was attempted by the group of Edward Rubin in collaboration with Pääbo, generating 65,000 nucleotides [22].Together, these results demonstrated that DNA sequences can indeed be generated from the Neanderthal nuclear genome, providing a proof-of-concept for further studies. Subsequent analyses indicated that the data sets generated by high-throughput sequencing suffered from contamination with modern human DNA [23], prompting further methodological refinements in the work that followed. In the final paragraph of his article [21], Pääbo stated that achieving one-fold coverage of the complete Neanderthal genome (3 billion base pairs) could be envisioned as further technical improvements were anticipated. Pääbo concluded by announcing that he had initiated a project aiming to achieve an initial draft version of the Neanderthal genome within two years.
A draft Neanderthal genome sequence
To fulfil his bold plan, Pääbo needed even more bone. Additional specimens from the Vindija collection were obtained in collaboration with the Berlin-Brandenburg Academy of Sciences and the Croatian Academy of Science and Arts. Three of these specimens, each about 40,000 years old, contained more than 1% Neanderthal DNA and were selected for sequencing of the Neanderthal nuclear genome. Additional material was obtained from other sources including Neandertal valley, Mezmaiskaya cave in Caucasus and El Sidrón cave in Spain. Specific adaptors were constructed and added under clean-room conditions, containing a Neanderthal-specific sequence key that unequivocally identified each sequence determined as derived from the extract of a Neanderthal bone [9], and additional critical methodological improvements were made. The protocol for library preparation was improved, resulting in several hundred-fold increased yield of readable DNA [24], and the ratio of Neanderthal to microbial DNA was increased by using restriction enzymes that preferentially cleave bacterial seq-uences in the libraries.
In 2008, the team started using emerging sequencing technology that allowed even higher throughput. They obtained one billion sequences of 30-70 nucleotides in length. The change of sequencing platform was challenging, as new algorithms needed to be developed for analyzing and mapping the data [25]. An alignment approach was implemented that accounted for the type of nucleotide misincorporation that dominates when analyzing ancient DNA (C to T transitions, particularly towards end of reads). The reference human and chimpanzee genomes and an inferred genome sequence of the common ancestor of humans and chimpanzees were used for com-parative analyses.
Different expertise was now needed for analysis of the data, including population geneticists, and a consortium of around 50 scientists was established. Amongst others, David Reich, Nick Patterson, Montgomery Slatkin and Jim Mullikin were critical contributors, as were Rasmus Nielsen and Weiwei Zhai who were later invited to the team. DNA from the three individuals resulted in sequence information encompassing more than 4 billion nucleotides, which were then mapped and analyzed. In 2008, the complete 16,565 nucleotide mtDNA was reconstructed using 8,341 mtDNA sequences. Analysis of the assembled sequence unequivocally established that the Neanderthal mtDNA fell outside the variation of extant human mtDNAs and allowed an estimation of the divergence between the two mtDNA lineages to 660,000 years [26].
In 2010, in a ground-breaking publication, Pääbo achieved the unachievable and reported a draft Neanderthal nuclear genome sequence [27]. His bold plan had been fulfilled. Five present-day Homo sapiens genomes from different parts of the world were also sequenced to aid in comparative data analyses. Three approaches were employed to estimate contamination with present-day human DNA in the data produced. First, positions known to differ between Neanderthal and present-day human mtDNA were used. Second, for bones derived from female Neanderthals, modern human male DNA contamination was estimated by looking for the presence of unique fragments from non-recombining parts of the Y chromosome. Finally, sites in the nuclear genome where present-day humans differ from both chimpanzee and Neanderthals were used. All three methods produced estimates of less than 1% conta-mination.
A smaller amount of DNA sequence was generated from Neanderthal bones excavated from three additional sites that cover much of the geographical range of late Neanderthals: El Sidròn in Asturias, Spain, Feldhofer Cave in Nean-dertal Valley, Germany and Mezmaiskaya Cave in the Caucasus, Russia. DNA divergences estimated for each of these specimens to the human reference genome showed that none of them differed significantly from the Vindija individuals. Thus, Neanderthals from across a great part of their range in western Eurasia seemed equally related to present-day humans. The average divergence of Neanderthal and present-day human nuclear DNA sequences was estimated to 825,000 years [27].
The question of whether interbreeding had occurred between Neanderthals and ana-tomically modern humans could now be investi-gated directly, by nuclear genome sequence ana-lyses. When Pääbo investigated how closely related Neanderthals were to present-day humans, initially using two European Americans, two East Asians, and four West Africans, he surprisingly found that Neanderthals were equally close to Europeans and East Asians, and signi-ficantly closer to non-Africans than to Africans. Extending the analyses by adding the genome sequences of French, Han, Papuan, Yoruba, and San individuals resulted in the same conclusion. The finding that Neanderthals were closer to the non-Africans than to the Africans was most easily explained by gene flow (introgression) between Neanderthals and the ancestors of non-Africans during their time of co-existence. As an independent indication of introgression, many genome regions with larger variation outside of Africa than within Africa were shown to represent Neanderthal sequences.
Neanderthals were as closely related to the Chinese and Papuan individuals as to the French individual, even though morphologically recognizable Neanderthals exist only in the record of Europe and western Asia. This may be explained by mixing of early Homo sapiens ancestral to present-day non-Africans with Neanderthals, likely in the Middle East before their expansion into Eurasia. The data suggested that between 1 and 4% of the genomes of people in Eurasia are derived from Neanderthals.
Further analysis identified variants that occurred in all sequenced present-day humans, while they were absent in Neanderthals. A total of 78 nucleotide substitutions that change the protein-coding capacity of genes were found to be fixed for the derived state in Homo sapiens, while Neanderthals carried the ancestral state. Relatively few amino acid changes have thus become fixed in the last few hundred thousand years of human evolution. Several potential non-coding regulatory substitutions were also found that were fixed in present-day humans compared to Neanderthals. The catalogue of unique human features has since expanded as genome sequence data has accumulated, forming the basis of a very active area of research.
Discovery of a novel hominin: The Denisovan
By sequencing the Neanderthal genome, Pääbo had demonstrated that the genome sequence from an extinct late Pleistocene hominin can be reliably recovered and analyzed, pioneering a new approach to study our evolutionary history. Exceptional circumstances are required for DNA to persist over the long time since our closest evolutionary relatives went extinct. Degradation increases with temperature and soil conditions such as acidity, and most bones from early hominins come from equatorial and tropical regions in Africa and Eurasia where conditions for maintaining DNA integrity are poor.
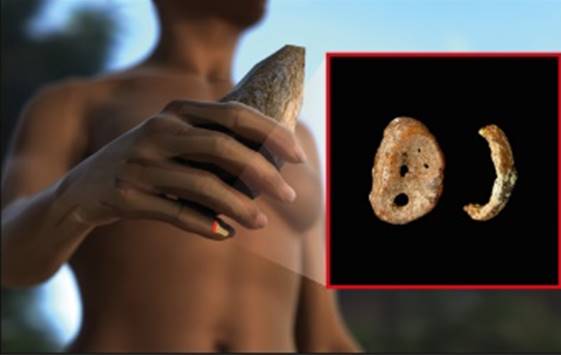
Figure 2. Distal phalynx of the hand, found in the Denisova cave.
However, archeological evidence indicated that archaic hominins also lived at higher latitudes, where the potential for DNA preservation is greater. One such region is the Altai mountains in southern Siberia where hominin occupation may have occurred more than 125,000 years ago. Bone specimens suitable for morphological classification do not exist from most sites in the Altai, but small pieces of human skeletons such as teeth and bone fragments have been recovered.
In 2008, the distal phalanx of the fifth manual digit of a juvenile hominin was excavated in Denisova Cave in the Altai mountains, Russia, in a stratum dated to 48,000 to 30,000 years ago. From this finger bone, Pääbo’s group made powder and extracted DNA, which was found to be exceptionally well preserved. Primer extension capture was initially used to isolate mtDNA fragments. These were sequenced and assem-bled to a complete mtDNA sequence with a mean coverage of 156-fold [28]. Several different approaches were used to assess the reliability of the mtDNA sequence. For example, a second DNA extract was used for shotgun sequencing, resulting in an assembled mtDNA sequence that was identical to the first sequence obtained by primer extension capture.
The sequence was aligned to 54 present-day human mtDNAs, a late Pleistocene mtDNA from an early modern human, six complete Neanderthal mtDNAs, one bonobo mtDNA and one chimpanzee mtDNA. Whereas Neanderthal mtDNA differs from that of Homo sapiens at an average of 202 nucleotide positions, the sample from the finger bone specimen differed at an average of 385 positions and the chimpanzee at 1,462 positions. Thus, the mtDNA from the unknown hominin was considerably more divergent from present day humans than from Neanderthal mtDNA. In 2010, a phylogenetic analysis of the sequences confirmed the astonishing fact that this was a unique hominin [28], which was named Denisova. The date of the most recent common mtDNA ancestor shared by the Denisova hominin, Neanderthals and modern humans was estimated to approximately one million years ago, or twice as deep as the most recent common mtDNA ancestor of modern humans and Neanderthals.
The implications of this finding were tremendous. Pääbo had discovered an entirely new hominin, distinct from Neanderthals and Homo sapiens. Furthermore, the discovery had been made entirely by retrieval and sequencing of archaic DNA, without any available morphological information. Pääbo’s team went on to sequence the nuclear genome from DNA extracted from the Denisova finger bone, following their strict protocols. They mapped the sequences to the human and chimpanzee reference genomes, as well as to the inferred ancestral genome of these species, generating a Denisova genome sequence with about 1.9-fold coverage [29].
The relationship of the Denisova genome to Neanderthals and modern humans was investi-gated. Whereas the team had shown that the divergence of the Denisova mtDNA to present-day human mtDNA was about twice as deep as that of Neanderthal mtDNA, the average divergence of the Denisova nuclear genome from present-day humans was similar but distinct to that of Neanderthals, indicating that they were sister groups. DNA sequences of the Neanderthals and the Denisova were estimated to have diverged on average 640,000 years ago, and from present-day Africans around 804,000 years ago.
Alignments were examined of sets of four genomes, one African, one Eurasian (French), one archaic hominin (Neanderthal or Denisova) and the chimpanzee. Both the Neanderthal and Denisova genomes matched the French genome to a higher degree than the African genome, but the archaic component of the Eurasian gene pool appeared less closely related to the Denisova individual than to Neanderthals. The fact that the Eurasian individual displayed greater similarity to the Denisova compared to the African individual suggested that Denisovans shared some of their history with Neanderthals before the gene flow from Neanderthals into non-Africans occurred.
Thus, the Denisova population did not appear to have been directly involved in the putative gene flow from Neanderthals into Eurasians. However, when the relationship of the Denisova genome to the genomes of 938 present-day humans from 53 populations who had been genotyped at 642,690 single nucleotide polymorphism positions was investigated, the Papuan and Bougainville islanders were distinct from almost all individuals.
This was explored further by analyzing additional genomes, supporting the conclusion that the Denisova individual contributed around 4-6% of its genetic material to the genomes of present-day Melanesians. This was surprising because it suggested that the geographical range of Denisovans was vast, spanning the Eastern and Southern parts of Eurasia. Based on these results, Pääbo and his team proposed that Denisova are a sister group of Neanderthals with a population divergence time of one-half to two-thirds of the time to the common ancestor of Neanderthals and humans. After the divergence of the Denisovans from the Neanderthals, there was gene flow from Neanderthals into the ancestors of present-day non-Africans. Later Denisovans and the ancestors of Melanesians mixed, which did not affect other non-African populations [29] (Figure 3).
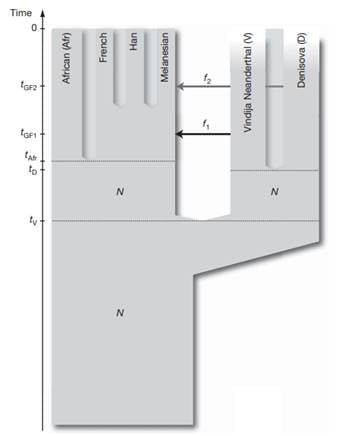
Figure 3. A model of population history compatible with the data. N de-notes effecttive population size, t denotes time of population sepa-ration, f denotes amount of gene flow and tGF denotes time of gene flow (Reich et al. 2010).
In 2012, Pääbo obtained a high-coverage (>30-fold) Denisova genome sequence from the original finger bone found in 2008, using a single-stranded library designed to increase the number of DNA molecules for sequencing [30]. In 2010, another hominin bone, this time a proximal toe phalanx, had been recovered in the Denisova Cave. In 2014, Pääbo’s team generated a complete, high quality genome sequence from this bone showing that it came from a Neanderthal individual from the Altai mountains [31]. This confirmed that the range of Neanderthals extended all the way to Siberia. They compared the genome to the available lower coverage Neanderthal genomes, including one from Mezmaiskaya Cave in the Caucasus and to 25 high coverage present day human genomes. Based on these data, >Pääbo’s group estimated the population split between Neanderthals and Denisovans to be 380,000 – 470,000 years ago and the split time between modern humans and Neanderthals/Denisovans to be 550,000 – 760,000 years ago. Further, they saw evidence of inbreeding in the Neanderthal lineage and very low levels of diversity, suggesting a small population size. They refined their estimate of Neanderthal ancestry in present-day Eurasian genomes to 1.5-2.1% and showed evidence for a more complicated history of admixture between Neanderthals and Homo sapiens. The team also found evidence of gene flow from Neanderthals in the Altai region into Denisovans.
An explosion in archaic hominin and ancient Homo sapiens genome se-quences
With his seminal discoveries, Pääbo established an entirely novel scientific discipline, now known as Paleogenomics. A wealth of genome sequence data has since been obtained from our extinct hominin relatives, as well as from ancient Homo sapiens, and the field is evolving rapidly. In 2014, Pääbo’s group used a hybridization approach to enrich DNA from the protein-coding fraction of the genomes of two Neanderthal individuals from Spain and Croatia and analyzed the sequences together with the Siberian Neanderthal genome. The genetic diversity of Neanderthals was shown to be lower and different from that of present-day humans, suggesting that Neanderthal populations were small and isolated from one another. It was further suggested that genes involved in skeletal morphology had changed more than expected on the Neanderthal evolutionary lineage whereas genes involved in pigmentation and brain function had changed more on the modern human lineage [32].
In 2017, Pääbo’s group sequenced a second Neanderthal genome at high coverage (>30-fold) from a Neanderthal dated to ≈50,000 years ago from Vindija Cave in Croatia [33]. She carried 1.6 differences per ten thousand base pairs between the two copies of her genome, fewer than present-day humans, supporting that Neanderthal populations were of small size. In 2018, Pääbo and Janet Kelso applied a method using hypochlorite treatment to generate low coverage (1-2.7-fold) genomic sequence from five Neanderthals who lived 39,000-47,000 years ago, referred to as late Neanderthals [34]. This doubled the number of Neanderthal genome sequences and enabled population genomic studies of Neanderthals. The group observed that the late Neanderthals resembled each other but were different from the older Neanderthal from the Caucasus, suggesting that the older Neanderthal lineages were replaced by a new population of Neanderthals. This population turnover likely occurred towards the end of Neanderthal popu-lation history.
In 2015, Pääbo’s group characterized the mtDNA genome and nuclear genome of two additional Denisovan individuals from the Denisova cave [35]. They showed that the nuclear genomes formed a clade with the original Denisovan genome whereas the mtDNA genome of one of the newly sequenced Denisovans was more divergent than the others, which they interpreted as indicating that the Denisovans were in the region for an extended period. In 2017, they sequenced the mtDNA and sequences spanning 47 Mb of the genome from a fourth Denisovan, concluding that this individual was substantially older than two of the other Denisovans, reinforcing the view that Denisovans were likely to have been present in the vicinity of Denisova Cave for a long time [36]. This was followed by a remarkable finding from a low coverage (2.6-fold) genome isolated from another bone fragment from the Denisovan cave [37]. Based on heterozygosity levels across the genome the group concluded that the individual was a first-generation offspring between a Neanderthal mother and Denisovan father.
Pääbo’s group subsequently published the third high-coverage Neanderthal genome [38], from an individual found in Chagyrskaya Cave in the Altai Mountains. She was more related to Neanderthals in western Eurasia than to Neanderthals who lived earlier in the Denisova Cave, consistent with the possible extinction of the older Neanderthal lineage and spread of later western Eurasian Neanderthals eastwards. Based on the numerous homozygous regions in this genome, they inferred that Siberian Neanderthals lived in relatively isolated populations of less than 60 individuals. Conversely, the Neanderthal from Europe, the Denisovan from the Altai Mountains, and ancient modern humans seemed to have lived in popu-lations of larger sizes.
In a particularly remarkable technological feat, Pääbo’s group recovered nuclear DNA sequences from two specimens from an assemblage of 28 hominin individuals, found in Sima de los Huesos in the Sierra de Atapuerca in Spain, which were dated to approximately 430,000 years ago [39]. These shared some morphological features with Neanderthals but their relationship to Neander-thals and Denisovans was not clear. Pääbo’s group showed that the Sima de los Huesos hominins were related to Neanderthals rather than to Denisovans.
In addition to Pääbo’s ground-breaking disco-veries of genomes from extinct hominins, the combination of an in-depth understanding of ancient DNA degradation, high throughput DNA sequencing technologies, and powerful compu-tational resources have led to the retrieval of ancient modern human genomes. An early example was the genome sequence of a 4000-year-old Eskimo, reported in 2010 from the group of Eske Willerslev [40]. In order to trace evidence of introgression from extinct hominins in early modern humans, Pääbo’s group generated genome sequences from a 45,000 year-old modern human from western Siberia [41] and a 40,000 year-old modern human from Romania [42]. Both showed evidence of recent intro-gression with Neanderthal genomes.
There is now a rich resource of genome data available from ancient human specimens found in vastly different geographical locations and from different time periods, and data are rapidly accumulating. The archaic genome sequences retrieved from our closest extinct relatives by Svante Pääbo have been used extensively in comparative analyses to shed light on population dispersals and human adaptive history. The scarcity of archaic hominin samples has limited the availability of genome sequences. Recently, Svante Pääbo, Matthias Meyer and colleagues showed that hominin mtDNA [43, 44] as well as nuclear DNA [45] can be recovered from Pleistocene sediments, opening entirely new possibilities to investigate the temporal and geographic distribution and population dynamics of extinct hominins. The high-coverage hominin genome sequences discovered by Pääbo constitute necessary scaffolds onto which less complete genome-wide sequence data from both sediments and bones can be mapped. The high-coverage genomes also represent a foundation for exciting and important comparative analyses with present-day humans.
Implications
Insights into our archaic evolutionary history
Pääbo’s discoveries have had a profound impact on the understanding of our evolutionary history, and they have galvanized research in the area. We now know that at least two distinct hominin groups, Neanderthals and Denisovans, inhabited Eurasia when anatomically modern humans (Homo sapiens) emerged from Africa. Three teeth have been found in Denisova Cave and shown by Pääbo’s team to carry genome sequences highly similar but distinct from that of the finger bone from which the original Denisova genome had been retrieved [29, 35, 36]. Thus, the teeth and the finger bone are likely derived from different individuals from the same hominin population. Morphological features were not shared with Neanderthal and modern human teeth, further indicating that Denisovans have an evolutionary history distinct from Neanderthals and modern humans.
The availability of genome sequences from our closest extinct relatives retrieved from varying geographical locations and timepoints have shed light on their distribution, population structure and admixture. Pääbo’s findings have been used to refine the Out of Africa model, in which all modern humans are traced back into Africa in an unbroken line. His data have revealed that interbreeding occurred between ancient modern humans, Neanderthals and Denisovans, resulting in small amounts of archaic genome segments retained in present-day humans. Non-African genomes contain around 1-2% Neanderthal genetic material. The locations of the introgressed archaic DNA segments vary between individuals, altogether around 40% of the Neanderthal genome has been found in present-day humans. Interestingly, some genomic regions completely lack signals from Neanderthal or Denisovan ancestry, referred to as “archaic deserts”, suggesting that introgression in these regions is not tolerated [46-48]. In addition to Neanderthal sequences, Melanesians received 4-6% of their genetic material from archaic Denisovans [49]. Multiple waves of introgression from Neanderthals and Denisovans to non-Africans have been indicated, reflecting many interbreeding events between ancient modern humans and extinct archaic lineages [48, 50]. Moreover, the proportion of Denisovan heritage differs markedly among Pacific groups [51].
Exceptional circumstances are required for DNA to survive in archaic bone and extinct hominin genome sequences have not yet been reported from Africa, despite this continent being the origin of our existence. Therefore, the contribution of archaic hominins to the genetic variation of present-day Africans remains less well understood. However, segments of archaic ancestry have been inferred without available reference archaic genomes and “ghost” archaic ancestry has been proposed in West African populations [52]. Neanderthal ancestry has also been detected in African individuals, a finding that can potentially be explained both by migration back to Africa from ancestral Europeans and gene flow into Neanderthals prior to their dispersal out of Africa. It was claimed that Neanderthal-like sequences can in fact be identified in every contemporary modern human genome analyzed to date [53].
One of the biggest remaining mysteries is why modern humans were so successful in their expansion and why the Neanderthals and Deni-sovans went extinct after having adapted to a Eurasian environment for several hundred thous-and years. The observation that these popu-lations were small, and that they had relatively high levels of inbreeding gives us a clue that they may not have been able to compete with the modern humans who were rapidly expanding in population size.
Relevance for human physiology
Svante Pääbo’s work provides exciting possibilities to search for archaic gene variants among present day humans and to study their association to phenotypes. As archaic humans were already genetically adapted for life in some of the environments of Eurasia, into which modern humans migrated, introgression events from Neanderthals and Denisovans could introduce alleles that positively impacted their ability to survive in the new environments after their expansion out of Africa. Introgressed sequences that have undergone positive selection can potentially provide functional information on parts of our genome, that is of general relevance for all living humans.
Segments that derive from Neanderthals can be found in most regions of the human genome, albeit at different frequencies in different parts of the genome. There is now strong evidence that some of these signals represent archaic haplotypes derived from introgression events. A clear example is the Denisova-derived version of the hypoxia pathway gene, EPAS1, which confers a genetic advantage to survival at high altitude and is found in present-day Tibetans [54]. The genomic region spanning the EPAS1 gene contains a set of SNPs that are found in the Denisova genome and in Tibetans, as well as at a very low frequency among Han Chinese. The boundaries of the haplotype-specific SNPs define a 32.7 kb region, a length that is consistent with the introgression event having occurred during the time when Denisovans and Homo sapiens co-existed. This haplotype structure is not observed in any other population group globally.
Another intriguing example of introgression is a cluster of genes encoding three Toll-like receptors, TLR6-TLR1-TLR10, known to be involved in microbial recognition and allergic reactions [55]. Janet Kelso and collaborators demonstrated that present day humans carry three distinct archaic haplotypes spanning these genes, indicating that several introgression events involving these genes occurred during our history of interactions with our archaic relatives. These haplotypes are present at variable, but surprisingly high frequencies in the current human populations, suggesting that they provide favorable biological effects. Additional examples of introgression events include a haplotype encoding the anti-viral signaling molecule Stat2 that is present at a high frequency in Melanesians [56], as well as haplotypes involving the 2′-5′-oligoadenylate synthetase (OAS) gene cluster, which also plays a role in anti-viral signaling, inherited from Neanderthals in a subset of Euroasians [57]. Recent studies by Pääbo and Zeberg demonstrated that archaic alleles on chromosomes 3 and 12, the latter involving OAS, can influence the risk for respiratory failure during SARS-CoV-2 infection [58, 59]. These and other studies suggest that much remains to be learned about the functional consequences of intro-gressed alleles for our physiology.
David Reich’s group, in collaboration with Pääbo, systematically inferred the extent of Neanderthal ancestry across the human genome and intersected a set of identified alleles with SNPs identified in genome-wide association studies to be linked to a set of disease-relevant phenotypes [46]. They also found that regions harboring a high frequency of Neanderthal alleles were enriched for genes affecting keratin filaments, as found by the group of Joshua Akey [48]. Databases such as the UK biobank have collected large numbers of phenotypes and genotypes, including variants that are likely introgressed from archaic hominins. Using this resource, Neanderthal-derived variants have been associated also with non-disease phenotypes such as pigmentation and sleeping patterns in present-day Europeans [60].
Additional examples exist of association between archaic alleles and human disease and non-disease phenotypes. When mixing with our archaic relatives, the ancestors of present-day humans encountered a reservoir of already adapted archaic alleles that could be useful as environments changed. Thus, archaic gene flow into Homo sapiens influences human physiology has occurred, offering exciting possibilities to elucidate how specific gene variants modulate biological processes at the molecular level.
This insight would not have been possible without the persistent work of Svante Pääbo and his team resulting in the definition not only of the Neanderthal nuclear genome, but also the unexpected discovery of a completely unknown hominin and the full characterization of its nuclear genome.
What makes us uniquely human?
Homo sapiens stands out among animal species by the complexity of our cultures, social structures, and our capacity to communicate. It can be assumed that this unique “humanness” results from changes in the genome on the modern human lineage. Access to archaic genomes offers exciting new possibilities to identify critical genetic features that distinguish us from archaic hominins. As our closest evolutionary relatives their genomes provide reference points that can elucidate specific human characteristics of rele-vance for physiology and medicine (Figure 4).
There are around 31,000 single-nucleotide positions in the genome where present-day humans from all parts of the world carry only a novel (derived) nucleotide, whereas both the Neanderthal and Denisovan genomes carry the ancestral nucleotide, conserved since the split from chimpanzee. Of the variants that are fixed in present-day humans, around 3,000 are in regulatory regions, around 30 affect putative splice sites, and around 100 affect protein-coding regions, altering amino acid composition [61]. Elucidating the functional relevance of uniquely human genetic variants is an exciting challenge of relevance to all Homo sapiens, which represents one of Svante Pääbo’s main current lines of research.

Figure 4. Archaic genomes offer exciting new possibilities to identify critical genetic features that distinguish modern humans from Neanderthals and Denisovans.
Gunilla Karlsson Hedestam, PhD, Professor at Karolinska Institutet (gunilla.karlsson.hedestam@ki.se), Member of the Nobel Committee
Anna Wedell, MD, PhD, Professor at Karolinska Institutet (anna.wedell@ki.se), Member of the Nobel Committee
Illustrations: Mattias Karlén
The Nobel Assembly, consisting of 50 professors at Karolinska Institutet, awards the Nobel Prize in Physiology or Medicine. Its Nobel Committee evaluates the nominations. Since 1901 the Nobel Prize has been awarded to scientists who have made the most important discoveries for the benefit of humankind.
Nobel Prize® is the registered trademark of the Nobel Foundation
References
- Cann, R.L., M. Stoneking, and A.C. Wilson, Mitochondrial DNA and human evolution. Nature, 1987. 325(6099): p. 31-6.
- Lander, E.S., et al., Initial sequencing and analysis of the human genome. Nature, 2001. 409(6822): p. 860-921.
- Venter, J.C., et al., The sequence of the human genome. Science, 2001. 291(5507): p. 1304-51.
- Nurk, S., et al., The complete sequence of a human genome. Science, 2022. 376(6588): p. 44-53.
- Genomes Project, C., et al., A map of human genome variation from population-scale sequencing. Nature, 2010. 467(7319): p. 1061-73.
- Mallick, S., et al., The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature, 2016. 538(7624): p. 201-206.
- Saiki, R.K., et al., Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science, 1985. 230(4732): p. 1350-4.
- Paabo, S. and A.C. Wilson, Polymerase chain reaction reveals cloning artefacts. Nature, 1988. 334(6181): p. 387-8.
- Briggs, A.W., et al., Patterns of damage in genomic DNA sequences from a Neandertal. Proc Natl Acad Sci U S A, 2007. 104(37): p. 14616-21.
- Hoss, M., et al., DNA damage and DNA sequence retrieval from ancient tissues. Nucleic Acids Res, 1996. 24(7): p. 1304-7.
- Paabo, S., Ancient DNA: extraction, characterization, molecular cloning, and enzymatic amplification. Proc Natl Acad Sci U S A, 1989. 86(6): p. 1939-43.
- Hoss, M. and S. Paabo, DNA extraction from Pleistocene bones by a silica-based purification method. Nucleic Acids Res, 1993. 21(16): p. 3913-4.
- Poinar, H.N., et al., Amino acid racemization and the preservation of ancient DNA. Science, 1996. 272(5263): p. 864-6.
- Lindahl, T., Instability and decay of the primary structure of DNA. Nature, 1993. 362(6422): p. 709-15.
- van der Valk, T., et al., Million-year-old DNA sheds light on the genomic history of mammoths. Nature, 2021. 591(7849): p. 265-269.
- Krings, M., et al., Neandertal DNA sequences and the origin of modern humans. Cell, 1997. 90(1): p. 19-30.
- Ovchinnikov, I.V., et al., Molecular analysis of Neanderthal DNA from the northern Caucasus. Nature, 2000. 404(6777): p. 490-3.
- Krings, M., et al., A view of Neandertal genetic diversity. Nat Genet, 2000. 26(2): p. 144-6.
- Serre, D., et al., No evidence of Neandertal mtDNA contribution to early modern humans. PLoS Biol, 2004. 2(3): p. E57.
- Margulies, M., et al., Genome sequencing in microfabricated high-density picolitre reactors. Nature, 2005. 437(7057): p. 376-80.
- Green, R.E., et al., Analysis of one million base pairs of Neanderthal DNA. Nature, 2006. 444(7117): p. 330-6.
- Noonan, J.P., et al., Sequencing and analysis of Neanderthal genomic DNA. Science, 2006. 314(5802): p. 1113-8.
- Wall, J.D. and S.K. Kim, Inconsistencies in Neanderthal genomic DNA sequences. PLoS Genet, 2007. 3(10): p. 1862-6.
- Maricic, T. and S. Paabo, Optimization of 454 sequencing library preparation from small amounts of DNA permits sequence determination of both DNA strands. Biotechniques, 2009. 46(1): p. 51-2, 54-7.
- Kircher, M., U. Stenzel, and J. Kelso, Improved base calling for the Illumina Genome Analyzer using machine learning strategies. Genome Biol, 2009. 10(8): p. R83.
- Green, R.E., et al., A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell, 2008. 134(3): p. 416-26.
- Green, R.E., et al., A draft sequence of the Neandertal genome. Science, 2010. 328(5979): p. 710-722.
- Krause, J., et al., The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature, 2010. 464(7290): p. 894-7.
- Reich, D., et al., Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature, 2010. 468(7327): p. 1053-60.
- Meyer, M., et al., A high-coverage genome sequence from an archaic Denisovan individual. Science, 2012. 338(6104): p. 222-6.
- Prufer, K., et al., The complete genome sequence of a Neanderthal from the Altai Mountains. Nature, 2014. 505(7481): p. 43-9.
- Castellano, S., et al., Patterns of coding variation in the complete exomes of three Neandertals. Proc Natl Acad Sci U S A, 2014. 111(18): p. 6666-71.
- Prufer, K., et al., A high-coverage Neandertal genome from Vindija Cave in Croatia. Science, 2017. 358(6363): p. 655-658.
- Hajdinjak, M., et al., Reconstructing the genetic history of late Neanderthals. Nature, 2018. 555(7698): p. 652-656.
- Sawyer, S., et al., Nuclear and mitochondrial DNA sequences from two Denisovan individuals. Proc Natl Acad Sci U S A, 2015. 112(51): p. 15696-700.
- Slon, V., et al., A fourth Denisovan individual. Sci Adv, 2017. 3(7): p. e1700186.
- Slon, V., et al., The genome of the offspring of a Neanderthal mother and a Denisovan father. Nature, 2018. 561(7721): p. 113-116.
- Mafessoni, F., et al., A high-coverage Neandertal genome from Chagyrskaya Cave. Proc Natl Acad Sci U S A, 2020. 117(26): p. 15132-15136.
- Meyer, M., et al., Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins. Nature, 2016. 531(7595): p. 504-7.
- Rasmussen, M., et al., Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature, 2010. 463(7282): p. 757-62.
- Fu, Q., et al., Genome sequence of a 45,000-year-old modern human from western Siberia. Nature, 2014. 514(7523): p. 445-9.
- Fu, Q., et al., An early modern human from Romania with a recent Neanderthal ancestor. Nature, 2015. 524(7564): p. 216-9.
- Slon, V., et al., Neandertal and Denisovan DNA from Pleistocene sediments. Science, 2017. 356(6338): p. 605-608.
- Zavala, E.I., et al., Pleistocene sediment DNA reveals hominin and faunal turnovers at Denisova Cave. Nature, 2021. 595(7867): p. 399-403.
- Vernot, B., et al., Unearthing Neanderthal population history using nuclear and mitochondrial DNA from cave sediments. Science, 2021. 372(6542).
- Sankararaman, S., et al., The genomic landscape of Neanderthal ancestry in present-day humans. Nature, 2014. 507(7492): p. 354-7.
- Schaefer, N.K., B. Shapiro, and R.E. Green, An ancestral recombination graph of human, Neanderthal, and Denisovan genomes. Sci Adv, 2021. 7(29).
- Vernot, B. and J.M. Akey, Resurrecting surviving Neandertal lineages from modern human genomes. Science, 2014. 343(6174): p. 1017-21.
- Reich, D., et al., Denisova admixture and the first modern human dispersals into Southeast Asia and Oceania. Am J Hum Genet, 2011. 89(4): p. 516-28.
- Villanea, F.A. and J.G. Schraiber, Multiple episodes of interbreeding between Neanderthal and modern humans. Nat Ecol Evol, 2019. 3(1): p. 39-44.
- Choin, J., et al., Genomic insights into population history and biological adaptation in Oceania. Nature, 2021. 592(7855): p. 583-589.
- Durvasula, A. and S. Sankararaman, Recovering signals of ghost archaic introgression in African populations. Sci Adv, 2020. 6(7): p. eaax5097.
- Chen, L., et al., Identifying and Interpreting Apparent Neanderthal Ancestry in African Individuals. Cell, 2020. 180(4): p. 677-687 e16.
- Huerta-Sanchez, E., et al., Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature, 2014. 512(7513): p. 194-7.
- Dannemann, M., A.M. Andres, and J. Kelso, Introgression of Neandertal- and Denisovan-like Haplotypes Contributes to Adaptive Variation in Human Toll-like Receptors. Am J Hum Genet, 2016. 98(1): p. 22-33.
- Mendez, F.L., J.C. Watkins, and M.F. Hammer, A haplotype at STAT2 Introgressed from neanderthals and serves as a candidate of positive selection in Papua New Guinea. Am J Hum Genet, 2012. 91(2): p. 265-74.
- Mendez, F.L., J.C. Watkins, and M.F. Hammer, Neandertal origin of genetic variation at the cluster of OAS immunity genes. Mol Biol Evol, 2013. 30(4): p. 798-801.
- Zeberg, H. and S. Paabo, The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature, 2020. 587(7835): p. 610-612.
- Zeberg, H. and S. Paabo, A genomic region associated with protection against severe COVID-19 is inherited from Neandertals. Proc Natl Acad Sci U S A, 2021. 118(9).
- Dannemann, M. and J. Kelso, The Contribution of Neanderthals to Phenotypic Variation in Modern Humans. Am J Hum Genet, 2017. 101(4): p. 578-589.
- Paabo, S., The human condition-a molecular approach. Cell, 2014. 157(1): p. 216-26.
Nobel Prizes and laureates
Six prizes were awarded for achievements that have conferred the greatest benefit to humankind. The 12 laureates' work and discoveries range from proteins' structures and machine learning to fighting for a world free of nuclear weapons.
See them all presented here.

